
Abstract
This paper reviews the prevailing myths regarding steam sterilization currently prevalent in the pharmaceutical industry. It updates an earlier article by the same authors on the that subject that was originally published in 1998. The text addresses the current situation with respect to the beliefs debunked in the original effort, as well as several new mistruths that have emerged in the intervening years.
Introduction
Nearly ten years ago, we published our original paper entitled “Moist Heat Sterilization—Myths and Realities” with the goal of clarifying some of the then common misunderstandings related to the practice of moist heat sterilization (1). We believed that there was a general lack of understanding on the part of the pharmaceutical industry relative to this important subject. Of greater concern was the fact that a troublingly unscientific approach to the development and validation of moist heat sterilization processes was rapidly becoming common industry practice. Additionally, regulatory inspectional comments, compliance opinion, and even international standards were seen to advance concepts which were not supported by evidentiary science.
We hoped that our article would provide some benefit to the many individuals we have encountered who are involved in sterilization cycle development and validation, but have not been afforded the opportunity to participate in moist heat sterilization courses, or gain direct laboratory experience with biological challenges. Also, we were then and are still concerned that the performance objectives that were being imposed on firms were not reflective of risk and in some cases ran counter to our own training and experience. We also feared that some practices, although done with the best of intentions and in compliance with an understanding of prevailing guidance, might actually be dangerous.
The reader might reasonably ask how the approaches that have evolved and the standards that have supported them could be dangerous, as they often lead to what could be considered ultra-conservative sterilization processes. The answer is that sterilization is inevitably a trade-off between anti-microbial effectiveness and material damage. The purpose of sterilization is not only to ensure microbiological safety, but also to preserve as completely as possible the usefulness of the material(s) being sterilized. Thus, a process that goes beyond what is required for microbiological safety at the risk of damage to materials that could risk sterility assurance or product safety in other ways is not a safe cycle.
We have seen many examples of overly aggressive sterilization cycles that actually produce inferior sterility assurance when the overall objectives of the process are considered. A very good example of an inappropriate sterilization process design is an autoclave cycle for elastomeric plugs or stoppers that result in stoppers which clump or misfeed due to damage to the material or lubricants and coatings used to assist in machineability. Other examples would be damage to valve seals or gaskets in a filling pump or time/pressure fill system and increased friability of polymeric tubing, increasing the risk of cracking or bursting. What these examples of material damage have in common is that they result in invasive interventions in aseptic processing. Thus, the oft-quoted maxims regarding sufficiency and balance in life apply very well to the science of sterilization.
The use of a bigger hammer approach, or what we have termed the “Butch Cassidy” approach to sterilization—the latter from the scene in the famous Hollywood movie in which Butch uses far too much dynamite to open a safe and, as money rains down from the sky, his accomplice the Sundance Kid dryly asks, “Think you used enough dynamite, Butch?”—can be dangerous, even though this may seem counterintuitive to the validation technician. Killing BIs without regard to the larger concern for overall sterility assurance providing an abundance of “overkill” can be inappropriate if it increases the potential for other failures. One might even think for a moment how absurd the notion of “overkill” is when thought about critically. Organisms, whether people or microorganisms, are able to die only once. This point bears emphasis because to many in our industry it appears that microorganisms die differently than complex multi-cellular organisms such as man. We hear the death of microorganisms referred to as a “logarithmic” phenomenon. Actually, at the single-organism level death isn't logarithmic. Organisms, be they microbes or people, are found in various conditions of health, but in terms of the attribute we call “life” there are but two states, dead or alive.
Consider for a moment the standard sterilization cycle of 121 °C for 15 minutes, which is still referenced in the European Pharmacopoeia and has been the most common laboratory cycle for decades (2). If this cycle really were standard it would preclude the possibility of many terminal sterilization processes. The real problem here is that this cycle really requires F0 values that approach 20 or more minutes, while many products are actually sterilized at F0's of 8 minutes or less. Dr. T. Sasaki of Japan's National Institute of Health suggested that parametric release might be considered for cycles that yield F0 > 2 minutes (3). If this idea seems shocking it is only because we fail to consider that typical microflora, even most spores, have D121 values of 0.2 minute or less. So the current perception that only cycles that yield relatively high F0 levels or kill a 106 population of our most resistant biological indicator (BI) are safe results in another unfortunate risk. The risk comes from not using terminal sterilization or post-aseptic fill heat treatments as often as we should. This is a result of standards that are too prescriptive rather than consideration of actual product degradation.
We have heard the opinion from the podium of symposia sponsored by Parenteral Drug Association (PDA) and other organizations that prescriptive standards are required because some firms are not staffed with high-level experts in critical disciplines. We do not share the opinion that highly prescriptive standards result in better sterile product safety, nor do we believe that prescriptive standards reduce the need for a firm to retain experts on staff.
Sterilization is a key expertise that should be required of any firm that manufactures sterile products. Suggesting that firms whose mission is to control microbial contamination ought not to have access to sterilization experts is very much akin to suggesting that we should be able to issue standards that would obviate the need for a hospital to have pediatricians, gynecologists, or surgeons. We wouldn't take our loved ones to a hospital that lacked specialized expertise just because it had purchased a “how to” book. For similar reasons the notion that we can avoid the need to have specialized expertise within our companies, or that we can provide “must do” standards that obviate the need for real expertise, is naïve.
We hoped that our original publication would foster an improved understanding of steam processing. The document had some initial impact, and perhaps with the recent publication of the PDA Technical Report #1 revision, greater insight will result (4). Nevertheless, we have updated our initial effort and renewed our exploration of the situation with respect to steam sterilization. Regrettably, most of the issues we addressed a decade ago are equally problematic today. While some of the problems we addressed in 1998 appear to have diminished, several others remain prevalent, and unfortunately at least three new myths have emerged.
This paper will briefly recap our earlier effort, with the same goal as originally stated. We hope not only to debunk some of the common (and unfortunately still prevalent) myths surrounding steam sterilization, but also to encourage readers to seek a greater understanding of the underlying principles of microbiology and thermodynamics before setting the acceptance criteria in their validation protocols. Most of all, we hope this revised look at common moist heat sterilization myths will encourage every firm that makes sterile products or relies upon sterilization processes for microbial control to ensure that its organization has the requisite expertise in the science of sterilization. Sterilization is a science and is knowledge-based; it is an unfortunate mistake to think that just anyone can develop and validate sterilization processes as long as they have detailed standards and guidance documents to follow.
Myth 1: The temperature of 121.1°C has great microbiological significance in terms of spore death.
Our earlier paper might have enlightened some about the arbitrary nature of 121.1°C as an essential for sterilization; however, very little has changed in the way of requirements. The reason for its persistence is complex and difficult to fully understand. We believe that inspectional emphasis on this specification and fears regarding “worst-case” validation are enough to ensure the continued concern about attaining 121.1°C during a steam sterilization process. However, as we stated in our previous paper on the subject of moist heat myths, the focus upon 121.1°C as an absolute specification is unnecessary and unfortunate. The insistence upon achievement of 121°C can make precise control of processes intended to yield an F0 of 8 or less more difficult than it ought to be. The emphasis on temperature attainment should be replaced by a focus upon total lethality delivery as measured in F0. It is given that for lethality to be a suitable measurement proper moist heat conditions within the process must be assured. However, this is no less true in the case of using 121.1°C or any other temperature as a target, because without assurance of true moist heat conditions the cycle will be at best inefficient and at worst unacceptable.
It appears to us that some of the mythology regarding 121°C has been dispelled. We are increasingly finding acceptance criteria of minimum F0 rather than minimum time-temperature in moist heat sterilization. This is encouraging, and we hope this trend continues, because all moist heat sterilization cycles should be defined by total lethality as expressed in F0 rather then merely time at a target temperature of 121°C. F0 is both a more accurate reflection of the actual anti-microbial effect yielded by a process and a far better means of comparing the effectiveness of one process to another.
It bears repeating though that 121.1°C is nothing more than the conversion of 250°F to the Celsius temperature scale. This temperature became standard for no reason other than it was a nice round number that correlates closely with 15 psig (2.05 Bar). There is no spore killing magic that starts at precisely 250°F. Perfectly efficacious sterilization cycles are run at lower temperatures, and much of the lethality that accumulates at lower temperatures can quite legitimately be included in the total accumulated F0 calculation.
Another unfortunate scientific oddity this focus on 121.1°C has created along with the “worst-case study” culture that has come to permeate the practice of validation is that validation technicians don't seem satisfied with 121.1°C because it is perceived quite wrongly as a minimum requirement. Therefore, actual target temperatures in sterilization practice tend to be a degree or so higher than 121.1°C. For example, a validation technician may target 121.5°C for validation and then increase the temperature target to 122.0 or 122.5°C for production cycles. The worst-case cycle thinking often includes a lengthening of the cycle for production as well, so that the cycle time used for validation increases by some factor in production. In our view at least some of this is attributable to the misperception that falling below 121.1°C at any point in a moist heat sterilization process equates with failure.
We have noticed over the last several years the emergence of what we might term Myth 1a. We have heard the opinion rendered that F0 is a scientifically invalid measure of moist heat sterilization lethality. The argument we've heard advanced in support of this anti-F0 position is that one could run a sterilization cycle for hours at temperatures of 60–70°C and find that cycle to be valid using a F0 calculation. Anyone who has spent even a few moments studying a F0 table or done calculations using the F0 equation will know this is simply not true. Accumulation of significant levels of F0 does not begin until bacterial spore-killing temperatures are reached.
The autoclave cycle that many people consider standard, which is 121°C for 15 minutes, is a standard microbiology laboratory cycle for soya bean casein digest media. In any basic course in bacteriology one learns that this cycle isn't applicable to all bacterial media. In fact some medically important, selective media must be sterilized at either lower temperatures or shorter exposure times. Really, there isn't or shouldn't be a standard autoclave cycle for anything. Sterilization is risk-based processing and the only standard that need apply is that of the minimum level of acceptable risk, which is currently defined as one non-sterile article per million. We have heard the famous John Sharp quote that this standard is just fine provided you “aren't the millionth bloke” (5) Mr. Sharp may have a point, but if one in a million isn't a suitable level of risk then let's have that discussion. It is a perfectly reasonable discussion to have, as you will see from Myth 2 which follows.
Myth 2: There is something very special about a sterility assurance level (SAL) of 10−6 because it defines the process capability of moist heat processing.
The recent and scientifically encouraging focus upon risk- and science-based regulation, which had its genesis in International Conference on Harmonization (ICH) Q9, may have helped in dispelling this myth (6, 7). It is now recognized that defining a SAL is nothing more than establishing a maximum amount of risk tolerance. One contaminated unit in a million was first defined by the food industry as a sterilization goal for canned foods (8). Its utility as a risk mitigation target for sterilization is a statement of the maximum risk that a firm will assume, only stated differently as the minimum process capability that is acceptable from a regulatory point of view. However, in the vast majority of processes, the process is substantially more lethal. Terminal sterilization of products typically results in SALs of better than 10−20; this includes cycles that yield F0s of 8 min or less. The SALs achieved in the sterilization of product contact materials can result in probabilities of ≤10−100 (9). When so-called “overkill” process designs are used, SALs of 10−300 or more are not uncommon. Obviously, when SAL values such as these are targeted, the objective must be something other than the abatement of risk. It should be obvious to anyone with even a basic understanding of exponents and statistics that the idea of a minimum acceptable process at a level of contamination risk of one in a million is effectively obsolete and without meaning. We suspect no one has ever seen a serious attempt to develop a moist heat sterilization cycle that actually yields a 10−6 SAL with a single log safety margin, which in arithmetic terms would be rather substantial as this would take the risk level to one in ten million. In fact, the multiple margins of safety in force in moist heat sterilization make an actual 10−7 process nearly impossible to realize. The actual risk levels utilized in our industry are substantially lower, as will be discussed below. In fact, they are so low that the 10−6 SAL has no real meaning in 2009, if indeed it ever had any real meaning. This is not to say that there haven't been sterilization failures, but when risk has occurred it has been related to things other than the lethality target. If one does not properly maintain or operate a sterilizer, then one has failed due to poor good manufacturing practice (GMP) compliance, not because of inadequate acceptance criteria. Brute force is no substitute for intelligent process design, GMP compliance, and proper attention to detail.
Myth 3: You need a biological indicator (BI) with a population of 106 to demonstrate a SAL of 10−6.
In several publications since the earlier myths effort, we have continued to debunk this myth. The BI is merely a safe and reliable means for the assessment of biological lethality in sterilization validation. Its destruction affirms a suitable agreement between biological lethality and the documented physical (thermometric and pressure) conditions of the process. The estimation of the SAL (or risk) for the process requires knowledge of the bioburden population and resistance and indicates what the probability of bioburden survival is in the routine sterilization process. The BI population is complementary to this determination, but there is no reason for its population to be fixed at 106 in order to be useful for its intended use. It follows that industry and the regulatory community could establish a new risk target, for example 10−8 without the need to use BIs at a population of 108.
By way of example, let us imagine that we have a product in which an inoculated BI has a D121 value of 2.0 minutes. Let us further assume that the maximum safe F0 value that can be used in the sterilization of this product is 12 minutes.
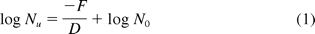 where
where
F—12 minutes
D—2.0 minutes
N0—106
Nu—Probability of a Non-Sterile Unit (PNSU)
PNSU—10−0 = 1
In this case the inoculation of 106 BIs per test article would result in a probability of BI survival of approximately 63%, something that we suspect many in our industry would consider a failure. However, let us make the quite reasonable assumption that the worst-case D121 of the most resistant recovered bioburden organisms was 0.2 minutes. This means that the very same 6 X D process that resulted in approximately a 6 log reduction of the BI in this example would in fact yield a 60 log reduction of the most resistant bioburden organisms. If we assume that the bioburden per unit is no more than 10 CFU and that the entire bioburden consists of the most resistant organism (a worst-case assumption), then the calculated SAL of each unit is 10−59. Thus the purpose of the BI is to demonstrate a general agreement allowing for biological variability between the biologically evaluated lethality and that measured thermometrically.
Finally, as described in the Myth 2 section, we as an industry have never really abided by a 10−6 SAL (PNSU) target in process design and validation. We've customarily chosen to exceed the minimum process requirements to further reduce risk. Also, since there is so much reliance on overkill process design, we typically ignore the true resistance and population of native microflora, without which an accurate calculation of a real-world SAL is impossible. So, one might properly ask why, since we don't really design to a 10−6 SAL target, should we consider a 106 spore population vital to our work, particularly since there was never a direct proportional relationship between absolute BI spore population and SAL in the first place?
Myth 4: Overkill means that you develop a cycle that gives a complete kill of BIs with a N0 of 106 and then you double that cycle.
That description actually describes the half-cycle approach, which is just what its name implies. The half-cycle approach is often mistakenly considered the only way to achieve overkill sterilization validation. The half-cycle approach has the benefit of simplicity, but little else in its favor relative to use for steam sterilization validation for many pharmaceutical products. Nearly every sterilization process is a compromise between conditions that will sterilize, and those that are destructive to the physical properties of the material or product being sterilized. The half-cycle approach is inappropriate for terminal sterilization and poorly suited for filters, stoppers, tubing, and other elastomeric materials where excess heat can have a deleterious effect on performance or use. Overkill should never entail a half-cycle approach for steam sterilization (9). Half-cycle sterilization processes evolved due to specific process control issues associated with ethylene oxide gas sterilization. The history as to why it came into use in steam sterilization, where process control parameters are easily measured, is unclear. Hopefully, in the future half-cycle cycle design will go back to being an approach that is used exclusively in conjunction with gas sterilization; it has no value in conjunction with moist heat sterilization.
Myth 5: The only BI organism that is valid for use in the testing of moist heat processes is Geobacillus stearothermophilus.
Organisms such as Bacillus subtilis, Bacillus coagulans and Clostridium sporogenes are frequently used to demonstrate the efficacy of moist heat sterilization processes, especially those used for the terminal sterilization of large volume parenterals. These organisms are entirely suitable and their use is well documented in the literature (10). Realistically, were there no concerns regarding regulatory acceptability, these indicators could find a wider usage in moist heat cycles design and validation. In fact, doing so was once a common and fully acceptable practice. The position stated in United States Pharmacopeia 〈1035〉 on BIs holds true: a BI is suitable for use when it provides a challenge to the sterilization process that exceeds the challenge of the natural microbial burden in or on the product (emphasis added) (11). Really, though, the most important use of BIs is to directly evaluate the biological lethality of a moist heat cycle as compared to thermal and steam pressure data. Any BI, regardless of species, that enables the sterilization scientist to make this assessment is acceptable for use. These organisms are now available commercially, which facilitates their use in sterilization validation.
Myth 6: BIs have been increasing in resistance over the last few years.
Reflecting on this issue, it was perhaps poor understanding of how BIs are to be used, rather than an actual increase in resistance, that prompted the inclusion of this myth. The process must deliver 9–10 times the labeled D-value for the BI organisms to be reproducibly destroyed in sterilization validation (assuming a 106 population in replicate studies with 20 BIs per run) (see Figure 1).

Microbial death curve showing BI challenge end point.
If the process doesn't achieve that, survivors are possible. Delivering a 6 log reduction to a BI with an initial population of 106 will result in 100 (or 1) viable spores. Therefore placing BIs with a population of 106 spores/strip in an autoclave where the delivered F0 is equal to 6 times the D-value of the BI is a recipe for failure if total kill of the BI is intended. Of course, it is possible to use BIs to evaluate biological lethality, and when used in this manner a fractional cycle approach actually provides accuracy of assessment that is never possible using a total kill approach.
Myth 7: Non-condensable gases and excessive condensate are major problems in autoclave steam supplies.
This myth has largely been dispelled; however, the difference in sterilization focus between US and UK practitioners remains. Most firms have chosen to switch rather than fight, and while the steam quality evaluation expectations of HTM-2010 have never been scientifically supported, they are still being enforced (12). In fact, it is important to note that real consensus has never been reached regarding just what specific attributes process steam should have for it to be considered of adequate quality. It is certainly understood that it should comply with pharmacopeial chemical quality standards. It is also understood that an engineering definition of steam quality for industrial processes existed long before the publication of HTM 10 (or HTM-2010) (13).
It is useful to consider what steam is as we consider steam quality. Steam is vapor phase H2O, just as H2O in the liquid phase is commonly known as water, and H2O in the solid phase is called ice. When vaporous H2O (steam) comes in contact with an object at a lower temperature it changes to water (condenses). It is not merely the energy transferred to an object by steam that produces lethality it is also the transfer of moisture to that object.
There is a direct and very important relationship between the temperature and the pressure of H20 for the vapor state to exist. At 121.1°C the pressure required for saturated water vapor is 15.2 PSIG (2.05 Bar); in absolute terms this is 29.9 PSIA under conditions where atmospheric pressure is 14.7 PSIA (1 Bar). At these pressure and temperature conditions water is said to exist as a saturated liquid. If we could isothermally maintain these conditions and increase the volume of the vessel without increasing the number of H2O molecules present, some of the water would vaporize to form steam. In this case both the liquid water present and the water vapor present (steam) would be at 121.1°C. Thus, at saturation a mixture exists of both vapor phase (steam) and liquid phase (water). Historically, the engineering definition of steam quality is nothing more than a term used to describe the amount of water vapor and liquid water in the mixture. This can be expressed by the very simple equation: X (steam quality) = Mass of Steam/Mass of Steam + Mass of Liquid Water. So a steam quality of zero would mean that no vapor is present, and that all water exists only as a saturated liquid. Young suggested that upon entry into the autoclave, steam with a quality of 97% would be adequate because it would minimize the “potential for wetting moisture absorptive items” (14). This steam would be said to have a dryness fraction of 3%. An engineering description of steam suggests that a 5% dryness fraction is an acceptable steam quality (15). The concern is not that 97% (or 95%) steam quality is necessary for microbial kill, but rather that packaging materials could be damaged by moisture. It is nearly impossible to supply truly dry steam although, in modern pharmaceutical steam supply systems containing moisture traps or separators, steam quality >95% is readily attainable and completely sufficient.
HTM-2010 added a further attribute, which the authors of that document defined as a component of steam quality, a limit on non-condensable gases. Actually, it is quite unlikely that non-condensable gases would be present in a steam supply line at a level that might actually affect sterilization efficacy. In fact, it is not particularly clear at what level non-condensable gases would interfere with sterilization efficacy. Nor is any documented scientific evidence available to support the tenets of HTM 2010.
Young wrote that from a microbiological perspective there are but three factors that affect lethality (14). These factors are
Time
Temperature
Moisture
When a large number of microorganisms are exposed to saturated steam at a fixed temperature, the percentage of survivors can be determined as a function of exposure time. Bigelow and others have reported that kill rates as evidenced by D-values increased nearly linearly as temperature increased (16). As a result of this observation he developed the well-known z-value concept. The third of these critical factors, moisture, is rarely discussed in the validation context yet it is far more critical than non-condensable gases. The effect of moisture can be seen quite clearly from the simple fact that it is necessary to run dry heat processes at far higher temperatures than moist heat processes to achieve the same result. Barker in 1933 reported that moisture was required to produce denaturation and coagulation of proteins (17). Lewith reported similar findings; thus it appears that moist heat kills by a different mechanism than dry heat, which is probably largely an oxidative process (18). Wang, Schorer, and Humphrey showed experimentally that activation energy and entropy changes for killing of spores and denaturing of protein are quite similar, which further supports the concept first suggested by Barker (19). It was further demonstrated in these studies and others that spores are quite permeable and freely exchange water with the environment.
Dry heat sterilization results in low water activity of spores and occurs at low relative humidity (RH). Dry heat sterilization is accomplished at an Aw = 0 (RH = 0). In steam sterilization the conditions are an Aw = 1 and the RH is 100%. Angelotti, and Murrell and Scott, showed that spore resistance actually increased from an Aw of 0 to 0.3 (20). They reported that D-value increases of up to 100 fold were possible at these reduced moisture levels. These data help explain the criticality of air removal. Since air and steam don't mix, poor air removal can result in low moisture at the sterilization target. Young points out that “all the air does not need to be removed from a sterilizer, but all surfaces requiring sterilization must be exposed to adequate moisture” (14). This is further reinforced by the fact that in some moist heat processes, air is purposely left in the chamber to minimize pressure gradients across products being sterilized. However, provided that the air/steam mixture is adequately uniform, something that can be assisted by a circulating fan, consistent sterilization is observed.
In our view, while air removal is vital it is pointless and even dangerous to exaggerate its importance. In fact, in sterilization air removal is merely a means to facilitate an end, and that end is a consistent moisture level across the load as accomplished by steam penetration. Similar to air, superheated steam adversely affects rate of kill because it also prevents transfer of moist heat to microorganisms.
While the global industry has largely acquiesced to UK demands in this area, we believe that the measurements required by HTM-2010 do very little to ensure good moist heat sterilization practices. The failure to remove a small quantity of air from a given package is nearly impossible to detect in a large vacuum displacement autoclave using a DART, Bowie-Dick, or other air removal instrument. This is why the use of BIs in the pharmaceutical industry in validation replaced the routine use of these air removal tests. Only the BI can provide evidence that temperature, time, and moisture were sufficient to meet pre-defined lethality targets. Residual air is problematic only to the extent that it prevents proper steam penetration from being achieved at the sterilization target, and this will manifest itself as spore survival.
We will reassert our original position: the bugs don't lie (21). Only a BI can demonstrate that the biological lethality confirms the expected physical lethality as estimated by temperature and pressure in a sterilization process. The BI provides the ability to analyze moisture the third critical factor, which physical tests alone cannot affirm and that is the presence of heat and moisture at the target location. Physical evidence alone is inadequate as the primary evaluation tool for sterilization.
Myth 8: Air in the autoclave can form an insulating layer around materials in the load.
We still see the occasional presentation citing this thermodynamically absurd notion. It violates so many natural laws; it is hard to imagine that it is still a belief (22). Specifically, the second law of thermodynamics, which concerns heat, entropy, and the direction in which thermodynamic processes occur, rules out this completely unproven (and unprovable) hypothesis of air forming an insulating layer around objects in an autoclave. The second law holds that heat does not spontaneously flow from a cold material to a hot material, but rather heat flows from a hot material (steam, for example) to a cold material (an object in a sterilizer load). In general terms, the second law states that in an isolated system—like an autoclave for example—concentrated energy disperses as a function of time, and as a result of this dispersion less concentrated energy is available to do work. Energy dispersal also means that differences in temperature, pressure, and density even out over time. Therefore, a useful generalization is that thermodynamic entropy is a measure of energy dispersal, and so the second law is closely connected with the concept of entropy. Also, as heat (energy) is transferred to air, the tendency of that air to diffuse is increased as a function of accelerated molecular activity. In 1905 Albert Einstein introduced a theory that was used by Jean Perrin to measure the movement of small particles in a stationary liquid demanded by the molecular-kinetic theory of heat (Perrin went on to win the Nobel Prize in Physics in 1926 for his work) (23, 24). The end result of this diffusion, which is sped up by heat (energy), is that all molecules within a container will eventually be evenly distributed throughout the container—which obviously means that the distribution of residual air primarily in the form N2 and O2 molecules (there will be some small quantity of residual air in any autoclave cycle) within an autoclave will move in the direction of uniformity and do so rather quickly at the elevated temperatures associated with autoclaving. Therefore, the entire idea that a layer of air would remain fixed as an insulating layer around an object is absurd both thermodynamically and in terms of diffusive molecular motion. We have jokingly offered anyone who believes in this myth the opportunity to sit in a gravity displacement autoclave wearing a scuba suit and allow us to turn on the steam. We doubt Einstein, Perrin, or Kelvin (one of the originators of the second law of thermodynamics), if they were alive today, would take us up on this offer.
Myth 9: If the autoclave doesn't have a steady-state temperature distribution range of less than 0.5°C, it must be replaced.
The origins of this lie primarily in Food and Drug Administration's (FDA's) 1976 Proposed Current Good Manufacturing Practices (CGMPs) for Large Volume Parenterals (LVPs), where concerns for over and under-processing fostered this expectation (25). Modern terminal sterilizers in our industry commonly meet this criterion. Extension to hard goods sterilization is unnecessary given the absence of meaningful adverse effects of heat, and the willingness of microorganism to die at a wide range of temperatures (albeit at differing rates). The real-world difficulties associated with meeting this requirement have largely dispelled this myth for parts sterilization without any assistance from us. Unmentioned in our earlier effort was the lack of clarity regarding what this impractical expectation intended. Does it mean all thermocouples (TC)s over the entire cycle duration? Or, does it mean all TCs excluding the first few minutes? Or, does it refer to all TCs over some undefined shorter period. Or, is it possible that it really means all TCs over a single time period? This myth was likely in decline before we mentioned it, and it hardly bears further discussion.
Myth 10: All autoclaves have clearly definable cold spots, which must be located.
This is a remnant of the FDA's proposed CGMP for LVPs, and was unfortunately further supported in PDA's initial monograph on steam sterilization (26, 27). It remains a concern for terminal sterilization (along with its exact opposite, the rarely mentioned hot spot) but is largely irrelevant in parts sterilization. The identification of cold spots as they correlate to individual load items is of real importance in parts sterilization and is a required activity and is termed component mapping or container mapping. In fact, given the criticality of moisture as a lethality factor, it is obvious that very small differences in temperature are irrelevant. Therefore, it is an exaggeration of physical reality to describe a location that is, for example, at a temperature which is 0.2°C lower than the mean as a “cold spot”. Obviously, such a minor difference is insignificant. Labeling such a location as “worst case” may in fact be a gross over-simplification of reality. Objectively, only a detectably different rate of kill can truly establish that a location is “worst case”. After all, temperature measurement without ensuring adequate levels of moisture cannot confirm the acceptability of a location within a load.
Myth 11: Fixed load patterns are required.
This myth applies only to terminal sterilization where load position can be important (largely during cooling of the load where small changes can dramatically alter the total lethality delivered upwards as well as downwards). In parts sterilization, many firms have validated sterilization processes with the items relocated in replicate runs to demonstrate that load patterns can be flexible.
We would be remiss if we didn't continue to remind the reader about the real importance of orientation and wrapping in sterilization. Sterilization wrapping materials must allow steam penetration and condensate removal, and their control is essential. Documentation of proper orientation for all load items is important as well to maintain and ensure consistency of air/condensate removal and steam penetration pathways in all loads.
Myth 12: Liquid water at 121°C is not as effective in sterilizing as steam at 121°C.
PDA's revised Technical Report No. 1 on steam sterilization includes information on the nearly identical heat capacity of steam and superheated water at the same temperature (28). Depending upon the process design, container, and the sterilizer, steam or superheated water can be successfully utilized either as a sterilization medium or as a source of energy to closed containers. Saturated steam is water in the vapor phase that is in equilibrium with its condensate, liquid water (29). Many terminal sterilization processes, especially those for plastic containers, rely on the superheated water for heat transfer to the containers. The interior of those same containers contains water in the formulation that is superheated during the terminal sterilization process. The key factors are that adequate temperature has been achieved in the presence of moisture. Whether the heat and moisture are provided by water vapor or superheated water is of no consequence. BIs immersed in water-filled containers are readily killed and yet are never in contact with steam.
Myth 13: The presence of superheated steam is a significant problem in contemporary steam sterilizers.
The attention paid to steam quality in the last decade resulted in the evaluation of superheated steam in many different locales. In the majority of instances we are aware of, there was never any difficulty in meeting this requirement nor did its momentary presence have a deleterious effect on process effectiveness in modern steam sterilizers that are used with properly designed steam supplies. Superheated steam, if it were to exist in a sterilizer, is a condition that would interfere with the attainment of proper moisture levels. Therefore, while temperature and pressure relationships are certainly important to monitor, the confirmation that superheated steam is not problematic is provided by the BI.
Myth 14: Physical data on sterilization cycles is inherently more reliable than the results of any microbiological testing.
Unfortunately this myth has survived largely unchanged since our prior publication. We suppose the ability to determine averages, minimum, and maximum temperatures, and plot the data from multiple points in color, lends physical data more credence than microbiological data. Spreadsheets, calibration reports, and statistics do not make it more reliable, however. Physical data provides a means to correlate the biological destruction in the sterilizer with the documentation provided by the sterilizer's control system on each cycle. Once the validation study is completed the documentation provides affirmation of the biological destruction. Given the choice of physical or biological data as confirmation of process lethality, assuming for a moment that only one of these methods existed, most trained sterilization scientists would prefer the biological result because it eliminates guesswork and integrates the three key factors required for effective sterilization. For those who place greater trust in physical data, consider the following:
A shotgun is accurately aimed at a 1-m diameter target 50 m away. At 50 m, the shotgun is expected to achieve a spread of 2 m. The shotgun is fired. Does one conclude that the target has been hit solely by virtue of the physical data, or does one go and examine the target to see if it has actually been struck? As scientists and engineers, we would probably all agree that checking the target would provide far more conclusive evidence.
The biological data provides the same clarity of evidence in sterilization setting. We should never rely on inference to determine the result of our experiments when more direct evidence is readily available. Consider that the physical data provides information on only two of the three important attributes for sterilization, temperature and time. Understand that only by adding to the analysis information from the BI can we really see the full effect on the target.
New Myths
Our initial effort addressed myths we had encountered numerous times prior to its publication; however, we were not so naïve as to believe that new myths would not emerge. The passage of time has brought to our attention three new myths of some prevalence. Regrettably, each of these has its origin in regulatory expectations.
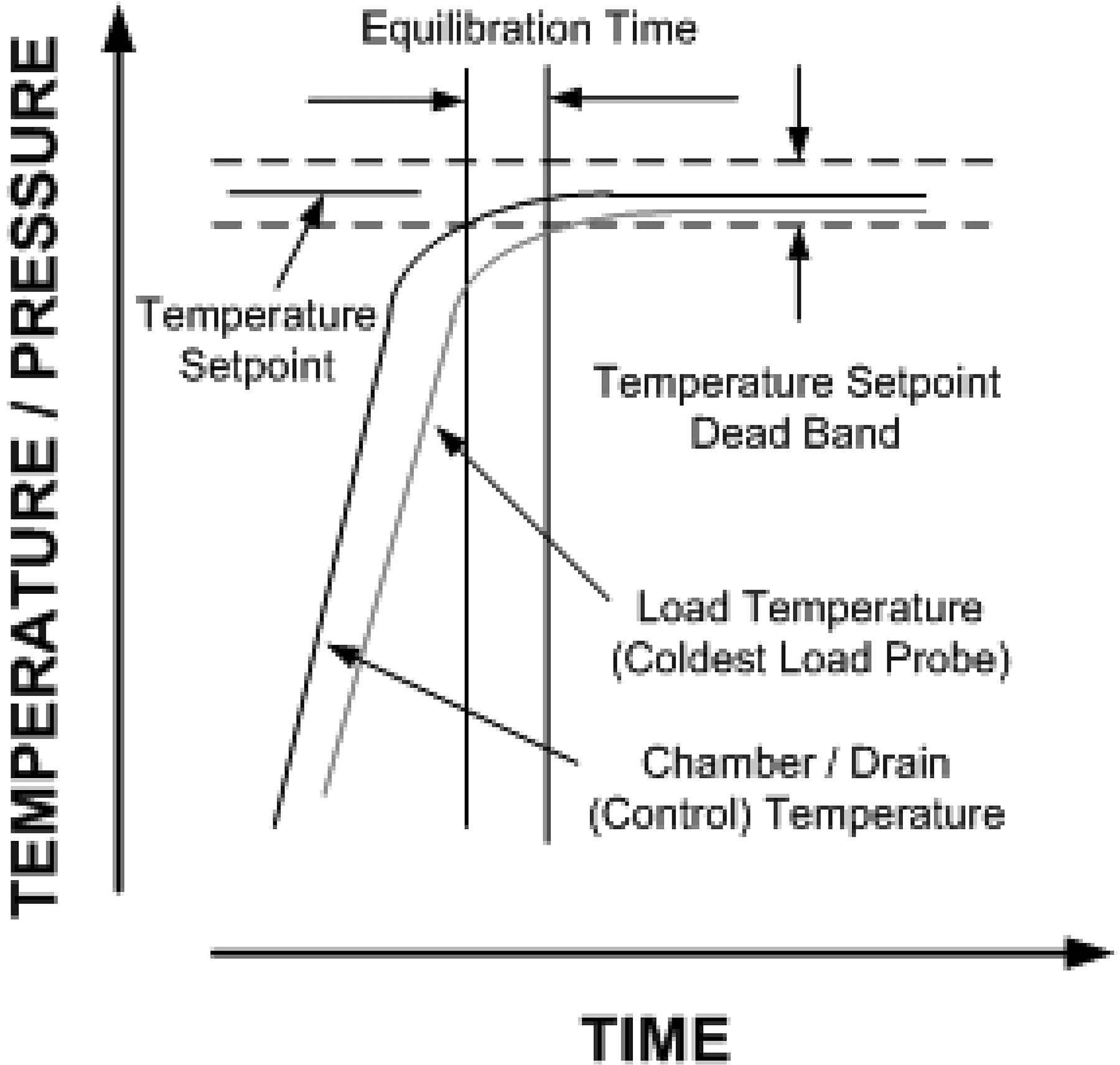
New Myth 15: Equilibration times must be less than 30 s.
This “requirement” for the time lag between start of the exposure period of the cycle and the slowest to heat penetration probe originated in HTM-2010, and was later incorporated into ISO 11134 and European Norm (EN) 285 (30, 31, 32) (see Figure 2).

Equilibration time.
Delays in come-up are presumed to confirm presence of air in the chamber, insulating the temperature probe, and are indicative of an air removal/steam penetration problem. The basis for the 30-s rule is unknown (as are many of the numerical “requirements” originating in HTM-2010). In small sterilizers, the expectation is that the equilibration time will be not more than 15 s.
Meeting this “requirement” has commonly been accomplished by increasing the number of pre-vacuums to as many as nine (the number required is a function of the depth of vacuum, size of the load, and mass of the items within it). Proponents argue that the increased vacuums enable more air to be removed. It is more likely that it is not the number of vacuums that resolve the “problem”, but rather that during the increased number of steam pulses items are heated closer to the exposure setpoint.
Reproducible come-up times have always been required in validation studies (the only activity where this time lag can be assessed), but are typically not more than 3–5 min. The time lag can be a natural consequence of the mass of the item adjacent to the temperature probe, and hence is expected by virtue of the laws of thermodynamics. Extensive prior history across the industry in thousands of validation efforts suggests the expected maximum equilibration time is extreme. The extensive success in gravity displacement autoclaves and sterilization-in-place (SIP) cycles where come-up times are substantially longer (due to the absence of forced air removal) was never considered. Thus industry has been presented with an unsupported performance constraint that is inconsistent with the historical facts and a large body of relevant data. As an aside we have started to see this expectation creeping into SIP processes where it has even less relevance and will likely force significant change to physical systems and sterilization methods for no meaningful gain.
New Myth 16: Determining the resistance of the BI on multiple substrates is a required part of the validation exercise.
The origin of this myth appears to be associated with difficulties with ethylene oxide sterilization and H2O2 decontamination of isolators. Resistance of BIs used for these processes varied depending on the substrate, for example, aluminum foil, stainless steel coupons, and paper/fiberglass spore strips. These differences have been attributed to penetrability of the different substrates. For H2O2, in many early efforts, when process expectation for this technology was uncertain, firms believed that the proper goal of these treatments was sterilization, rather than decontamination. Perhaps because H2O2 was believed to have limited penetration, D-value studies were initiated with spores dried on the various surface comprising the isolator and load (33). The results were variable, but within the same range of values. Where the BI on its well controlled substrate material was reported to have a D-value of 4 min at a specified condition, the load materials evidenced above and below this number, but always in the same order of magnitude.
This experience manifested itself in steam sterilization of elastomeric closures where expectations for D-value became commonplace in conjunction with new product submissions. The experience with steam sterilization has mimicked that with H2O2, as the D-values reported for the elastomeric materials vary above and below the reported D-value of the spore itself, but typically in the same order of magnitude. The FDA offered the following in its 2004 aseptic guidance:
“It also should be noted that the resistance of microorganisms can vary widely depending on the material to be sterilized. For this reason, careful consideration should be given during sterilization validation to the nature or type of material chosen as the carrier of the biological indicator to ensure an appropriately representative study.” (34)
This perspective rejects long-standing validation practices in which BIs are utilized as “worst-case” surrogates for the bioburden. Destruction of the bioindicator demonstrates that sterilizing conditions have been attained, or that targeted biological lethality levels have been reached. The real confidence in sterilization is established by the substantial difference in resistance between the indicator spores and the ordinary bioburden microorganisms. As the spores can be 1,000,000 times more resistant than the bioburden in steam sterilization, the routine process is overwhelmingly safe. If the process is altered to accommodate varying resistance because of presumed substrate concerns, the lethality delivered to the bioburden will be increased beyond what are already extremely safe levels.
It should be remembered that in routine sterilization the BI microorganism is not present; only the bioburden microorganisms are (see Figure 3).

Comparative death curves of BI and bioburden.
The substantial safety factors we have with steam sterilization because of the extreme resistance of the preferred BI means little in the context of patient safety. BIs are tools that we use to evaluate sterilization cycles; they should not be used to define a sterilization cycle. That is a risk-based exercise that should be the province of sterilization experts within an organization.
New Myth 17: The only good BI is a dead BI.
There is the persistent belief that BI survivors are to be avoided at all costs in validation exercises. The notion is that their survival casts suspicion on the lethality of the cycle. To those conversant in sterilization, this notion is clearly wrong. One of the common means to establish the resistance of the biological challenge microorganism is the survivor curve, where the reduction in population is plotted against the time of exposure to the process (see Figure 4).

Microbial death curve kill zones.
Where the BI is fully inactivated, the true lethality of the process is less certain. The microbial challenge might have died in the first minute or last minute of the exposure period, which would represent substantial differences in the cycle's lethality against an indicator microorganism of known resistance. The presence of survivors from the resistant BI at the end of the exposure period is the only way to accurately define the lethality of the process. The death of all of the BI organisms during the validation, while conceptually desirable, actually provides less information about the process being evaluated. Those who might doubt the veracity of this are encouraged to revisit any standard sterilization text (35, 36, 37, 38).
Fresh Perspectives on the Steam Sterilization Myths
There are unfortunately several misconceptions about steam sterilization still prevalent in the industry. Our efforts in this area have improved the situation somewhat; however, many of the myths persist. Newer regulatory input has clearly not helped the situation. The emergence of new myths regarding steam sterilization is most troubling. This requires a reminder to those who would set performance standards and to those who declare compliance to those standards, that is, those who could negatively affect sterilization practice. The reminder is simply this: well intentioned entities and individuals must refrain from recommending or endorsing any standard that is not supported by scientific fact. The definition of scientific fact is quite simple. A scientific fact can only result when a clearly stated hypothesis is verified by experimental data resulting from well-designed, well-controlled, and fully documented scientific experiments. In science we have a formal way in which experimental results become scientific facts. This requires the duplication of the experiments in multiple laboratories to ensure reproducibility and then the careful scrutiny of those results by peers. Only then does a hypothesis become an established scientific fact.
Moist heat sterilization has been a subject of scientific inquiry and experimentation for over a century. A very rich literature exists and verifiable scientific facts abound. It follows then that any standard proposed should follow established scientific fact. The hypothetical has no place in any industry standard. Mere opinion is an anathema to the standard setting process. We ask those who have the important responsibility for proposing standards to hold their work up to the highest standards of scientific integrity and rigor. Steam sterilization has proven to be an extremely robust process in our industry, and our struggles to adhere to arbitrary dictates and overly conservative viewpoints serve little purpose. If we are to properly consider risk in pharmaceutical operations, surely there must be processes other than moist heat sterilization that deserve attention. Aseptic processing, manual sanitization, and aseptic gowning are areas where fuller consideration of the inherent uncertainties might provide real benefits to patient safety. The preoccupation with steam sterilization on the part of many in this industry is inappropriate if it results in reduced attention to other, more significant risks to sterility assurance.
Simplifying a technology in hopes of diminishing the need for in-depth knowledge is tempting; however, we believe that in the long term it is wrong and counterproductive. Rather than proposing standards that are hoped to avoid the need for scientific sophistication, we should be making sure that those whose mission is to sterilize health care products are knowledgeable and understand the science that supports moist heat sterilization technology.
Footnotes
- © PDA, Inc. 2009
References




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Expanding the Use of Moist Heat for Terminal Sterilization
- The Relation Between the Load, Duration, and Steam Penetration Capacity of a Surface Steam Sterilization Process: A Case Study
- A Risk-Based Approach to Variable Load Configuration Validation in Steam Sterilization: Application of PDA Technical Report 1 Load Equivalence Topic