
Abstract
An emerging trend in the biotechnology industry is the utilization of plastic components in manufacturing systems for the production of an active pharmaceutical ingredient (API) or a finished drug product (FDP). If the API, the FDP, or any solution used to generate them (for example, process streams such as media, buffers, and the like) come in contact with a plastic at any time during the manufacturing process, there is the potential that substances leached from the plastic may accumulate in the API or FDP, affecting safety and/or efficacy. In this article the author develops a terminology that addresses process streams associated with the manufacturing process. Additionally, the article outlines the safety assessment process for manufacturing systems, specifically addressing the topics of risk management and the role of compendial testing. Finally, the proper use of vendor-supplied extractables information is considered.
LAY ABSTRACT: Manufacturing suites used to produce biopharmaceuticals can include components that are made out of plastics. Thus it is possible that substances could leach out of the plastics and into manufacturing solutions, and it is further possible that such leachables could accumulate in the pharmaceutical product. In this article, the author develops a terminology that addresses process streams associated with the manufacturing process. Additionally, the author proposes a process by which the impact on product safety of such leached substances can be assessed.
Introduction
An emerging trend in the biotechnology industry is the utilization of plastic components in manufacturing systems for the production of an active pharmaceutical ingredient (API) or a finished drug product (FDP) dosage form. This utilization of plastic components in the place of traditional manufacturing equipment is driven by economic and quality considerations. On the economic side, the use of disposable plastic technology may reduce the expense associated with the construction and maintenance of the manufacturing suite. For example, a plastic component in a manufacturing suite may be discarded after use, thus eliminating the need for traditional cleaning and cleaning validation activities. From a quality perspective, plastic components may be more compatible with materials used in or encountered during the manufacturing process than are either glass or metal (e.g., the oxidation or inactivation of an API or FDP by trace elements leached from metal production equipment). Examples of such plastic disposables include (1)
Tubing and connectors used to transfer starting buffers into the process stream
Syringes used to transfer liquids
Filters used with starting solutions, process intermediates, and/or the final API
Bags (and associated tubing) used to store and transfer process-related solutions
Tangential-flow filters used for concentration and diafiltration of process intermediates
Chromatography resins and membranes used for the concentration and/or purification of process intermediates
O-rings used to seal connectors and sanitary fittings
Final bulk containers
Sensors or other process-facilitating devices
If the API, the FDP, or any material or solution used to generate them (for example, process streams such as media, buffers, and the like) come in contact with a plastic at any time during the API or drug product manufacturing process, there is the potential that substances leached from the plastic may accumulate in the API or FDP. In this very general respect, then, plastic components of manufacturing systems are no different than plastic packaging (container closure) systems. However, while packaging situations involve direct and final contact between the plastic and the API or FDP, manufacturing situations may be much less clear-cut in terms of the nature of the plastic/API or plastic/FDP interaction. For example, if the contact between the manufacturing equipment and a solution occurs early in the manufacturing process, the solution may undergo significant additional processing before it actually becomes the final product. In fact, it is possible to envision numerous manufacturing situations in which the contacted solution and the API (or FDP) are not directly related, in the sense that the solution (or its components) does not directly become the API (or FDP). In cases of such a remote and indirect contact, there is some question whether substances extracted from the manufacturing equipment during the contact persist in the fully processed API or FDP. This uncertainty related to the potential impact of the interaction between the API or FDP and its manufacturing system translates into uncertainty in terms of the proper studies to perform to efficiently and effectively establish and assess the magnitude of any API (or FDP) safety issue related to the interaction.
It is well recognized in the pharmaceutical industry that interactions between an API or FDP and its manufacturing system must be controlled and minimized to ensure that the API or FDP is suitable for its intended use. The general requirements for manufacturing systems are contained in the regulations of several international regulatory agencies. In the United States, the requirements are enumerated in Title 21 of the Code of Federal Regulations, Part 211.65, which states that “equipment shall be constructed so that surfaces that contact components, in-process materials, or drug products shall not be reactive, additive, or absorptive so as to alter the safety, identity, strength, quality, or purity of the drug product beyond the official or other established requirements” (2). In the European Union (EU), a related statement is found in the EU Good Manufacturing Practice document, which states that “production equipment should not present any hazards to the product. The parts of the production that come into contact with the product must not be reactive, additive or absorptive to such an extent that it will affect the quality of the product and thus present any hazard” (3). Additional guidance for APIs can be found in the ICH Q7 GMP Guidelines, which state that “equipment should be constructed so that surfaces that contact raw materials, intermediates or APIs do not alter the quality of the intermediates or APIs beyond the official or other established specifications” (4).
While all potential interactions between manufacturing equipment and the API or FDP are important in terms of the suitability of use of the API or FDP, this article focuses on additive processes, and specifically those processes in which an entity from the manufacturing system migrates into, and becomes entrained in, the API or FDP. Clearly such an added entity is itself an impurity whose direct and indirect effect on the safety and efficacy of the API or FDP must be established. Although the regulations make it clear that additive interactions must be considered, there is no detailed regulatory guidance on how to specifically address and assess additive interactions between the API (or FDP) and its associated manufacturing equipment. Additionally, while all aspects of suitability for use must be considered in evaluating manufacturing systems, the focus of this article is specifically on safety aspects.
As the use of plastic materials in manufacturing systems has grown, so too has the concern that such materials may affect the quality or suitability of use of the API or FDP. As a result of such growing concerns, terminology and strategies for impact assessment have been developed and proposed (1, 5–11). As greater experience and understanding is obtained in terms of the nature and magnitude of manufacturing system API or FDP interactions, it is reasonable that such proposals be reviewed and modified as appropriate. Thus the purpose of this article is to propose and discuss a nomenclature related to such interactions, to discuss a general outline of the safety assessment process for manufacturing systems, and to consider the proper use of vendor-supplied extractables data.
Terminology
Evolution and Utilization of Terminology for Packaging Systems
The development and adoption of a standardized nomenclature and the enumeration of key concepts establishes the basis for effective communication, which will greatly facilitate the ability to address the often subtle and complex idiosyncrasies of the safety assessment process. Considering an appropriate nomenclature to describe substances that start in one entity (for example, a plastic material) and end up in a different entity (for example, a drug product) due to contact that occurs between the two, it would be useful if the nomenclature established (a) where the substance originated and (b) where the substance ultimately resides. Furthermore, it is useful if the nomenclature is intuitive in the sense that the distinctions made by the nomenclature are clear and unambiguous. Thus, when the pharmaceutical industry began to grapple with the compatibility issues associated with drug products and their packaging systems, the terms leachables and extractables emerged and were adopted into common usage. Initially, these were synonymous terms used interchangeably. However, it was quickly established that not only was the interaction between a packaging system and the packaged drug product complex and multifaceted but also that there existed multiple methods to measure or determine the magnitude of the interaction. It therefore was recognized that these two similar terms could be distinctly defined so as to provide clarity, consistency, and focus to the compatibility and/or safety assessment process.
It is readily understood in the scientific community devoted to compatibility assessment that establishing the magnitude of the interaction between a drug product and its packaging system can be accomplished in two ways: by testing the packaging system (or its materials or components of construction) or by testing the drug product. Specifically, one could definitively establish the exact magnitude of interaction if one discovered and measured substances that had leached into the drug product. However, as the drug products can be difficult to characterize and the leached substances are typically present in small quantities, an alternate, more analytically viable approach could be envisioned. In such an indirect approach, substances would be extracted from the packaging system (or materials or components thereof) using analytically expedient solvents, and the solvents would be characterized for the extracted substances, which may be present in the extracts at higher levels than in the drug product. The resulting information could then be used to infer the magnitude of the drug product–packaging system interaction and to serve as a guide for the development of appropriate analytical methodologies for the detection and quantitation of leached substances in the drug product.
In this scenario, there is a need to differentiate between those substances that are present in an extract of the packaging system and those substances that are present in the drug product itself. Conveniently, the prefixes “extrac-” and “leach-” effectively serve this purpose and thus the similar but distinct terms extractables and leachables were established (Table I), where an extractable is a substance that can be extracted from a packaging system component and which could end up in a drug product, and a leachable is a substance that is present and measured in the drug product because it has leached from the packaging system. These definitions are consistent and equivalent to those that have appeared in various regulatory guidelines and industry best demonstrated practice recommendations (Table II).
Derivation of the Existing Terms for Packaging (Container-Closure) Systems
Definitions for Extractables and Leachables
As the complementary terms extractables and leachables have become more commonly (and more correctly) used, it is clear that the existence of these terms facilitates a consideration of many issues related to compatibility assessment. For example, the strict and proper use of the terms allows for a very clear delineation of the roles and responsibilities of the two major players associated with FDPs, those being the vendor of the drug product itself and the supplier of the packaging (or more commonly the materials from which the packaging system are constructed). As extractables are a trait of a material that is measured by testing that material, it is clear that extractables testing could be the responsibility of the material's supplier. Alternatively, as leachables are a trait of the drug product that is measured by testing the drug product, it is clear that leachables testing is the responsibility of the drug product vendor.
Similarly, the two terms can clarify the proper testing strategy to be used when considering or assessing the impact of changes to the packaging system. In the simplest sense, an approved and marketed packaging system could experience two types of changes during its time in the marketplace. The first of these changes is the “natural” lot-to-lot variation in raw materials, manufacturing processes, etc. The issue related to these changes is one of quality control (QC), that is, are the raw materials and/or manufacturing processes under tight enough control that an acceptable output (in this case an effective and safe drug product) is consistently generated? The second of these changes is the “unnatural” interruption in the procurement of approved materials that occurs when the material's supplier makes a change to that material; for example, the supplier wants to change the material's formulation or production process. The issue related to these types of changes is one of change control (CC), that is, does the change in the packaging system affect the safety of the packaged API or FDP?
At first glance, it may seem that ongoing QC and CC are two closely related, subtle variations on the same theme, as they both deal with maintaining the quality of the drug product in its post-approval production batches. In actuality, however, they differ greatly in concept and context. Ongoing QC addresses the question, “Does a particular batch of incoming raw material meet a defined specification?” As an ongoing activity throughout the product's market lifetime, QC of incoming raw materials is most effectively addressed by testing the raw material itself and not by testing the finished product. Thus, in the case of raw materials used in packaging systems, ongoing QC is an exercise that involves extractables testing. In the case of CC (i.e., dealing with materials supplier–initiated changes in the composition or production process of a raw material), the essential issue at hand is “How does this change affect the compatibility aspects (specifically safety and/or efficacy) of an approved construct-product combination?” Because ultimately safety and efficacy are addressed via a migration study, CC is an exercise that involves leachables testing. Thus the distinction between issues of QC and CC is clarified by the distinction between extractables and leachables, and proper utilization of these terms dictates the type of strategy used to address either issue.
As a third point, there are any number of reasons, other than safety assessments why a plastic material or a drug product would need to be characterized with respect to its composition. An example of one such reason would be compliance of the drug product with compendia requirements. It is frequently the case that the compositional analysis of a plastic material involves the solubilization of some or all of the test material as a prerequisite to chemical analysis. If the solubilization of the material leaves the bulk of the material intact and removes only its additives and minor constituents, then the solubilization is more correctly termed an extraction, and a substance in the extract is correctly termed an extractable. Alternatively, the characterization of a drug product for its impurities could result in the delineation of leachables if the source of the impurity was the drug product's packaging system. Thus the terms extractables and leachables can be consistently and appropriately applied to the process of characterizing plastic materials and drug products.
Finally, the terms both properly establish the specific nature of drug product safety assessments. Because safety assessments based on leachables data involve analysis of the actual drug product, they truly reflect the actual clinical exposure and establish the actual clinical risk. Because safety assessments based on extractables do not involve the analysis of the actual drug product, they do not reflect the actual clinical exposure and thus only estimate the actual clinical risk. In fact, it is the general expectation that a safety assessment based on extractables data establishes the worst-case patient exposure to potential leachables. The degree to which this expectation is met depends on the similarity, or differences, between the extraction conditions and the actual product use conditions. In any event, a nomenclature that differentiates between extractables and leachables is a nomenclature that clarifies the difference between a worst-case and a real-case safety assessment and supports the proper utilization of such assessments.
Extension of the Terminology to Include Manufacturing Systems
Extending the issue of product-plastic contact from packaging systems to manufacturing systems adds to the complexity of the compatibility assessment because a third entity is present in the system. That is to say, a packaged product is manifested in two entities, the package and the packaged product. As noted previously, extractables testing involves testing the packaging and leachables testing involves testing the packaged product. In the case of manufacturing, there are three entities of interest: the manufacturing system itself (as a source of extractables), the manufactured product (which could contain leachables), and process streams, where process streams are solutions in the manufacturing process that either are converted to the manufactured product by the manufacturing process (intermediates) or facilitate the manufacturing process (e.g., cleaning solutions, diluents, elution buffers).
It is clear that when process streams contact plastic materials in the manufacturing process, entities from the plastic materials can migrate into the process streams. Thus there is at least the possibility that these migrating substances could persist during manufacturing and become leachables in the FDP. The question then becomes, “What term does one use when describing these migrating substances in a process stream?” Clearly these substances do not fit the definition of leachables, which are directly related only to the finished product. Additionally, these substances do not fit the definition of extractables because they are not obtained by extracting contact materials with solvents under laboratory conditions.
In order to extend the existing terminology to the case of a process stream, a new prefix and a new suffix are needed. Thus the prefix “migr-” is added to reflect “substances that are present in a process steam due to its contact with a material.” Similarly, the suffix “-ant” is added to reflect “a substance originating from a manufacturing system and/or its associated materials and components.” These additions produce the term migrant, which is defined as a substance present in a manufacturing process stream that is derived from the API's or FDP's manufacturing system, where a manufacturing process stream is a solution that is further processed, in whole or in part, to produce the API or FDP. It is noted that the term migrant and its definition are consistent with terminology proposed by the Bio-Process Systems Alliance, BPSA (18).
Risk Management and Safety Assessment
The utilization of risk management principles and concepts to establish and manage the product safety risk associated with contact between the product and its associated packaging and/or manufacturing systems is a cornerstone of global regulatory and industry thinking related to safety assessment. Industrial scientists and regulators agree that the concepts and principles of risk management have a definite strategic role in terms of designing, implementing, and interpreting effective and efficient impact assessments with respect to extractables, leachables, and their related manufacturing counterparts. In point of fact, regulatory guidance for container/closure systems makes very clear reference to, and very extensive use of, risk assessment processes and procedures (for example, Table 1 in the FDA Container Closure Guidance, reference 1, reproduced here as Table III).
Examples of Packaging Concerns for Common Classes of Drug Productsb
The successful utilization of risk management in safety assessment depends on the strategic and tactical manner in which it is applied. As safety assessment is a process and not a single action, the primary strategic issue related to the application of risk management in safety assessment is how and when risk management is applied in the process. From a tactical perspective, the major issue in the use of risk assessment is tied to the specifics of the risk assessment model itself. It is observed that these two issues are closely related, as it is the combination of a valid strategy and effective tactics that produces the desired outcome (i.e., achieving an acceptable level of risk).
Greatly over-simplified, the safety assessment process is one in which the dose or patient exposure to a specific entity is compared to an amount (or threshold) of that entity that has been determined to represent an acceptable safety risk (i.e., probable lack of an adverse impact on patient health and well-being). Proper risk management is practiced on the threshold side of this equation via the conservative application of sound toxicological assessment principles to relevant and credible toxicity data. Proper risk management is practiced on the dose side of the equation by producing sufficient information to establish the anticipated exposure.
There are essentially two means to experimentally determining the anticipated dose: an extractables (or migrants) evaluation and a leachables evaluation, where the extractables evaluation results in an inferred dose and the leachables evaluation results in an actual dose. It is the nature of extractables assessments that the inferred dose is the maximum dose. Furthermore, it is the nature of extractables and leachables assessments that the former is generally easier to perform. Thus a viable strategy for safety assessment is a two-step process involving an extractables assessment and then a leachables assessment. Between the extractables and leachables assessments is a decision point, where the decision point addresses the question, “Is the extractables assessment sufficiently rigorous and complete to establish that an acceptable level of risk exists?” If the answer to this question is “yes,” then no further leachables testing is necessary to assess the safety risk.
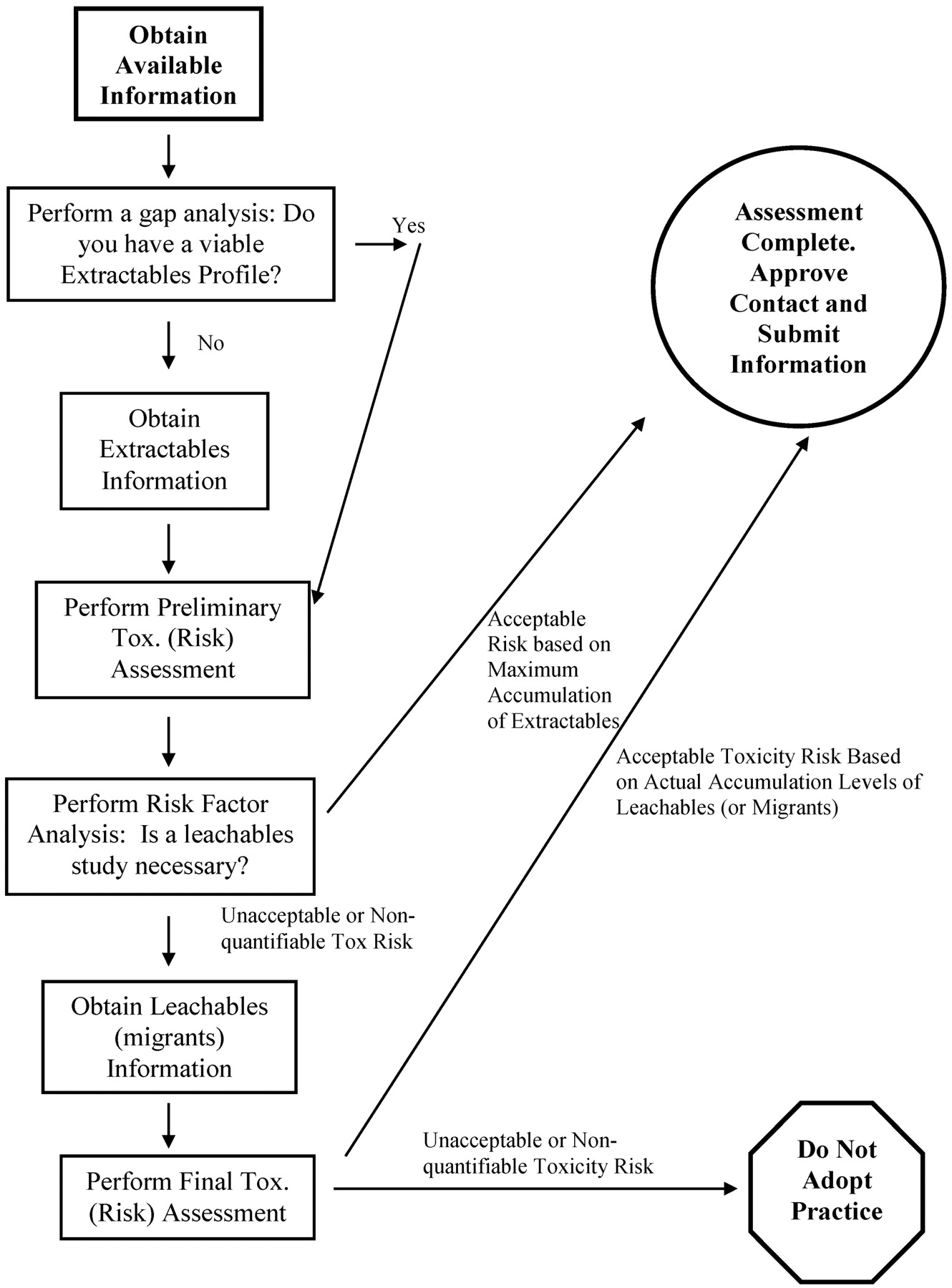
Such a risk assessment process is shown in Figure 1. Because extractables information on materials, components, or even entire systems could possibly be obtained without the API or FDP vendor having to perform extraction studies (by obtaining extractables information from the material's supplier, for example), the first step of the process is information collection and gap analysis. Gaps in an information-generated extractables profile would need to be filled in by extraction studies. Once a viable extractables profile has been obtained, the safety risk assessment process can be initiated with a preliminary toxicological assessment. The outcome of such an assessment, tempered with an assessment of the nature of the contact situation, is considered in the risk factor analysis. The risk factor analysis specifically addresses the question, “Can the safety assessment be completed on the basis of extractables information alone or is a leachables study necessary?” Factors to be included in this risk factor analysis will be considered in greater detail later in this article. If the answer to the risk factor analysis is that the toxicological risk can be shown to be acceptable based on extractables information only (that is, based on inferred worst-case risk), then the risk assessment process is completed with the approval of the contact situation. If leachables information is required to establish the actual risk, then the leachables study is completed and a final toxicological assessment is performed. The final toxicological assessment can have only two outcomes: either the final toxicological assessment concludes that there is an acceptable safety risk, in which case the contact situation is approved, or the assessment concludes that there is an unacceptable safety risk, in which case the contact situation is rejected.
It is noted that this concept of establishing acceptable risk for manufacturing systems is completely consistent with an analogous concept for packaging systems. For example, Table 1 of the FDA Container Closure Guidance, which is reproduced as Table III in this article, is an example of a two-dimensional risk management matrix for use in the safety assessment of packaging systems. In this particular table, the dimensions that establish the degree of concern (risk) that a safety-impacting interaction will occur between a product and its packaging are the route of administration of the product and the likelihood for an interaction to occur. Depending on where specific dosage forms fall in this grid, the dosage forms are classified as having lowest to highest risk with respect to a safety-impacting interaction. Such a classification has a direct and clear impact on the type and magnitude of testing that would have to be performed in order to properly establish and manage the risk of any safety-impacting interaction.
It is clear that there would be considerable value in the development of a similar risk assessment tool for manufacturing systems. In the context of Figure 1, this tool would be used to perform the risk factor analysis. Because of the complexity of the manufacturing process, it is clear that such a tool would have to consider multiple dimensions and establish the aggregate impact produced by all the individual dimensions.

Flow diagram illustrating the general risk assessment process for manufacturing systems. Sufficient information is obtained to delineate an extractables profile, which is then subjected to a preliminary toxicological safety assessment. Depending on the outcome of this assessment, the contact is either deemed to be safe or an additional investigation (leachables study) is performed.
Such a tool, referred to in terms such as risk evaluation worksheet (19) and risk assessment matrix (20), has been proposed by numerous authors (1, 5, 10, 19–21). Although the exact form and content of these devices differ somewhat among the various authors, the devices share a common theme, as illustrated in the generalized risk evaluation matrix shown in Table IV. In such an approach, several contributors to risk are identified and used to construct the matrix. These contributors may include things such as
The nature of the material itself and its inherent “resistance to extraction” or “tendency to possess extractable substances” or “tendency of the extractables to be toxic”
The nature of the solution itself and its “extraction capability”
Conditions of contact such as duration, contact surface area, temperature of contact
Proximity of the contact solution to the FDP
Generalized Risk Evaluation Matrix
In order to assess the safety risk using the risk evaluation matrix, the specifics of the contact situation are weighted via a ranking scale. Thus for example, the risk reflected in contact duration might be weighted as “less than 24 h = low = 1,” “greater than 24 h but 30 days or less = medium = 5,” and “greater than 30 days = high = 10.” Similar weightings would be given for the other contributors to the total risk. The final individual risk contributions are added up and a final risk score is obtained. The magnitude of the final risk score would dictate whether extractables and/or leachables testing would be performed. If the risk score were sufficiently low, one could conclude that such testing would not be necessary in order to establish acceptable risk.
If it is possible to support the concept of the risk management matrix but to have misgivings about proposed risk management matrices themselves, then this is the position of this author. This author notes that while a risk management matrix has the foundations of good science because (a) it is a process and (b) it involves a calculation, there is reason to be concerned with the potentially arbitrary and nonscience-based aspects of the matrix. Questions about the matrix that are open to scientific debate include:
Are all the relevant dimensions captured in the matrix?
Are the risk values used in each dimension science-based? In most situations, the published risk factors appear to be intuitive, experience-based, or arbitrary.
Are all the dimensions properly weighted? In most situations the various dimensions are equally weighted in terms of their impact on safety.
Is the aggregate effect of the dimensions additive (which is the process used in most applications) or are the interrelationships between dimensions more appropriately expressed by a more complicated function?
Are risk scores properly calibrated? For example, how has one determined that a risk score of 15 is safe and a risk score of 60 is unsafe?
What are the criteria or tests necessary to establish the qualitative levels within each risk variable? For example, how does one establish whether a material is reactive, interactive, or inert?
Given the great diversity of manufacturing operations, is it possible that a single matrix can be adopted that will “cover all the bases”?
It is this author's assertion that unless the specifics of risk evaluation matrices can be scientifically justified, the matrices are, at best, a qualitative tool that provides only a general or approximate answer to the question of whether a leachables assessment is necessary to obtain a rigorous safety assessment of manufacturing systems. However, it is not necessarily fair and proper for this author to challenge the scientific foundation of the risk assessment matrix as being subjective without at least considering the question, “Where will the information come from that would make the matrix more rigorously science-based?” In fact, the answer to the question can be found in the previously noted example of packaging systems, specifically those associated with orally inhaled and nasal drug products (OINDPs). When faced with the challenge of addressing such issues as “How low does one go in extractables and leachables assessments?” and “What are the science-driven best demonstrated practices for performing extractables and leachables studies?,” industry leaders in analytical chemistry and toxicology, supported by various other stakeholders, performed the analyses and studies necessary to produce the scientific information from which appropriate and justifiable answers could be obtained. On the packaging side, these efforts continue as various groups look to extend the answers obtained for OINDPs to other dosage forms (such as parenterals and ophthalmics). It seems to this author that such a model for information generation and interpretation could be extended to the world of manufacturing systems to address issues uniquely relevant to such systems.
Furthermore, it is this author's assertion that the risk assessment matrix contains redundant features when it is applied to the risk assessment process after an extractables assessment has been performed and thus that the matrix can be greatly simplified. For example, note that Table IV includes risk variables such as extraction strength of the solution, contact duration, contact surface area, and contact temperature. Such factors were considered and addressed in the design of the extractables assessment. Thus such factors are redundant when using the risk evaluation matrix between the extractables and leachables steps and they can be removed. Similarly, the material type risk variable is redundant because the extractables data itself provides for a quantitative means of establishing the reactivity of the material under evaluation.
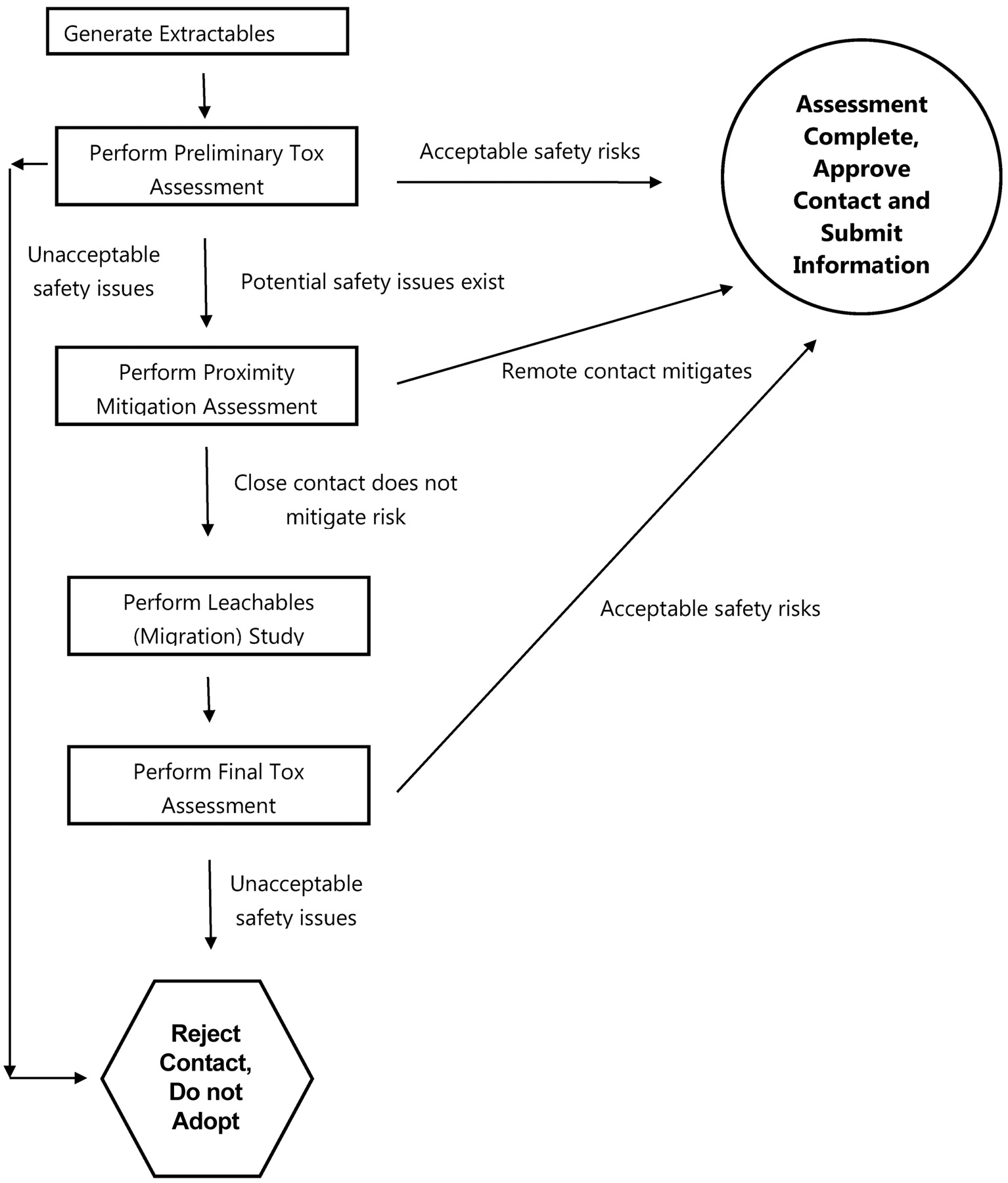
Thus an optimized risk evaluation matrix applied to address the question “Is an extractables study suitable for risk assessment or must a leachables study be performed?” has only two dimensions: the outcome of the preliminary toxicological assessment and the proximity to the finished product (Table V). The utilization of the optimized matrix is not a mathematical process of trying to estimate relative risk using various qualitative “factors”; rather, it is a two-step logical process with only two possible outcomes (Figure 2). The first step of the process is to consider the results of the preliminary toxicological assessment (based on the extractables data). If the results of this preliminary toxicological assessment are that the worst-case safety risk is acceptable, then no further consideration is necessary and the contact situation is approved. If the results of the preliminary toxicological assessment are that a potential safety risk exists, then one asks, “Does the proximity to the finished product mitigate the risk?” If the answer to this question is yes (as might be the case, for example, if the contact occurs in the very early stages of a multistage manufacturing process), then a leachables assessment is not deemed to be necessary and the contact situation is approved. If the answer to this question is no (or don't know for sure, as might be the case when the contact occurs in the last manufacturing step), then a leachables assessment must be performed in order to establish the safety of the contact situation. As noted in Figure 1, the results of the leachables assessment are subjected to toxicological evaluation to determine the final status of the contact situation.
Optimized Risk Evaluation Matrix

Flow diagram illustrating risk assessment process for manufacturing systems based on extractables toxicity assessment and proximity risk mitigation. The combination of the extractables toxicological assessment and proximity risk mitigation analysis dictates whether leachables testing is required to demonstrate contact safety.
It is a cornerstone of this article that the risk evaluation matrix is used to potentially eliminate the need for leachables testing. In some cases, it has been proposed by other researchers that the risk evaluation matrix can be used to eliminate even extractables testing. This is not appropriate for the fundamental reason that an extractables profile is the minimum information with which to perform a toxicological assessment. A risk evaluation matrix cannot be used to eliminate extractables testing because the information that is produced by extractables testing, that is, the identity of the extractables and their doses, is the essential basis of a toxicological assessment. Risk management without a science-based toxicological assessment is not proper risk management, because without a toxicological assessment the process of risk assessment becomes qualitative and subjective.
The concept of extractables information as a necessary foundation of the toxicological assessment is in complete agreement with the fundamentals of risk management. Specifically, a fundamental relationship in risk management is within the definition of risk itself, which is expressed as
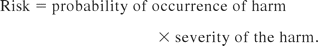
The risk evaluation matrix addresses only one term in this equation: the probability of the occurrence of harm. Thus by itself, the risk evaluation matrix does not allow for proper risk management. It is the extractables testing, coupled with its associated toxicological assessment that provides the severity of the harm term. Thus it is the combination of the extractables data and the risk evaluation matrix that allows one to assess risk and determine whether additional risk evaluation actions (e.g., leachables testing) are necessary.
In some cases risk evaluation matrices have been based on test methods that incorrectly have been concluded to produce “extractables-like” information. Thus, for example, a risk evaluation matrix may use compendial testing (such as USP 〈661〉) or general chemical test methods (such as nonvolatile residue or total organic carbon or UV absorbance) to address the reactivity or general safety of the material. This author notes that such a risk evaluation matrix is flawed, as this type of testing does not produce the compound-specific type of information (identity and concentration) required for safety risk assessment. The fact that such compendial testing would even be construed as “extractables testing” is based on an incomplete and less-than-rigorous utilization of the terminology (see Table VI). Compendial testing superficially resembles extractables testing because compendial testing involves the generation of an extract and the testing of the extract. Compendial testing is not extractables testing because
The extractions used in the compendia are generally standardized and typically do not mimic the conditions of actual contact between the system and the solution of pharmaceutical interest.
Compendial testing does not produce the compound-specific identities and concentration estimates that are critical parts of an extractables assessment.
Based on the combination of points 1 and 2, compendial testing does not produce data that can be utilized to assess toxicological safety risks.
Comparison of Compendial Testing versus Extractables Testing
Testing Intervals in a Migration (Leachables) Study
An important design consideration in a migration study is the number and timing of test intervals. The use of multiple time points in a migration (leachables) study performed with primary packaging is standard and expected practice. Such a strategy is predicated on the observation that the final drug product and its packaging system are in constant and direct contact over shelf life. Such constant and direct contact at least offers the possibility of increased migration of leachables over time and is the reason that the migration study for packaging must have multiple time points.
This is not the case for leachables associated with manufacturing processes. The absolute amount of a leachable that is present in the drug product due to the manufacturing process is established once the manufacturing process is completed and contact between the drug product and the manufacturing equipment has ended. Subsequent storage of the drug product in its final packaging cannot increase the concentration of a manufacturing related leachable. Thus the maximum concentration of a manufacturing-related leachable in a drug product is greatest at the earliest shelf life testing interval, which is commonly known as time zero. The practical ramification of this observation is that migration studies for manufacturing leachables consists of only one time point, and that time point is time zero.
Furthermore, it is observed that the ideal test article for a leachables assessment is not the packaged final drug product but rather the drug product sampled at the point of its filling into the final packaging. This is the case because there is a reasonable expectation that as a result of filling, the final drug product may contain two types of extraneous compounds: leachables from the manufacturing process and leachables from the final packaging. As it is at least theoretically possible that the presence of packaging leachables will adversely affect the accurate quantitation of manufacturing leachables, it follows that the ideal sample to test for manufacturing leachables in the unpackaged (but fully processed) FDP.
Proper Use of Suppliers' Extractables Data
A question that is frequently asked by users of manufacturing system components and systems is, “Can one use supplier-provided extractables information as the sole basis of a safety assessment?” The answer to such a question is clear when one considers the requirements for extractables data that are used in safety assessment. Specifically, there are two critical aspects of an extractables study: the generation of the extract and the testing of the extract. Each will be considered separately.
The regulatory guidance for packaging systems is clear on the topic on how to generate an extract. The extraction solvent is a medium that has the same propensity to extract as the drug product but is used in place of the drug product because it is easier to analyze. Extraction conditions such as time and temperature of contact are adjusted to accelerate (but not unnecessarily exaggerate) the exact clinical conditions of use experienced by the drug product. The same guidance is applicable to manufacturing systems as well.
It is often the case that what a supplier typically supplies as extractables information is a standardized extraction approach designed to be applicable for a large number of potential users of their plastic (or component or system). The very fact that a supplier's plastic/component/system could be used in numerous user applications means some applications will be less rigorous (more gentle) than the standard extraction conditions used by the vendor and some may be more harsh. The larger the difference between the standardized extraction conditions used by a supplier and the customized extraction conditions that are applicable for a particular user, the lesser the likelihood that the supplier's extractables information is applicable to the user's specific situation and thus the greater the likelihood that a safety assessment based on the supplier's data would be inappropriate for the user's situation. Thus it is clear that a user would only accept the supplier's extractables information if the extraction process used by the supplier was a reasonable simulation of the user's conditions of contact.
Even if the supplier produced the right extract, there is another issue that must be considered before the user accepts the supplier's extractables data. Specifically, the user must carefully consider the analytical process and methods used by the supplier to characterize the extract. Such a consideration must recognize the three primary objectives of the characterization process: find all the extractables, identify all the relevant extractables, and obtain an accurate assessment of the concentration of the relevant extractables in the extract. A fourth desirable but optional objective is to offer proof that these activities have in fact been accomplished.
A potential user of supplier data must review and examine, in great detail, the supplier's analytical methods. This review must go well beyond the typical “alphabet soup” approach, which might sound something like, “OK, they used GC/MS, and thus it must be good data.” The review must address all aspects of the analytical process, including any sample preparation steps, with great emphasis on the issues of scope (i.e., how many compounds would the method fail to respond to?), detectability (how low can you go with the method?), and accuracy (how confident is one in the concentration estimates that are generated?). An outcome of a poorly designed or implemented analytical process is a flawed safety assessment. In the extreme case, it is noted that there is no surer way to get a “didn't find any” extractables profile than to use methods that would never have found anything in the first place.
If, as a result of a rigorous assessment of the supplier's methods for generating and testing an extract, a user concludes that a supplier's study is both applicable and rigorous, then there would be no issue with taking the supplier's study results and using them as the starting point in a user's toxicological safety assessment. Even in the case that the supplier's study is found to be lacking and thus cannot be the basis of a user's safety assessment, any and all information provided by the supplier should facilitate the user's own extractables assessment.
Conclusion
A nomenclature has been developed that expands the concepts of extractables and leachables to manufacturing systems by introducing a new term, migrant. This term, coupled with extractables and leachables for packaging systems, represents a nomenclature that unambiguously establishes the origin and residence of all substances relevant to a consideration of the interaction that can occur between drug products (and/or their related solutions) and either packaging systems or manufacturing systems. A process for assessing the safety/toxicological risk associated with contact between drug products and manufacturing systems has been developed and is based on the requirement that extractables testing is a minimum requirement regardless of the anticipated risk. The utilization of risk evaluation tools and compendia testing in the safety assessment process is discussed. This author suggests that the utilization of risk assessment matrices be limited to those cases where the dimensions of, and processes used by, the matrices are established with, and justified by, good science. Additionally, the use of compendial testing in safety risk assessment is not recommended, as such testing does not produce the type of information upon which a safety assessment can be based. It is observed that a migration study of manufacturing system leachables consists of a single testing interval, specifically that interval typically called time zero. Finally, it is recommended extractables information obtained from a plastic material's supplier can be used by a user as the basis of a safety assessment only in the case that the user has ascertained that the extraction conditions used are appropriate for the user's application and that the analytical processes used for extract characterization were rigorous, complete, and appropriately sensitive.
Acknowledgment
The author thanks Jerold Martin, senior vice president of Pall and president of BioProcess Systems Alliance (BPSA), for his review of the article and his many insightful comments and recommendations. It is noted that the fact of this review does not imply that the views expressed in this article are, or reflect, those of Mr. Martin, of Pall, or of the BPSA.
Footnotes
-
Conflict of Interest Declaration The author declares that he has no competing interests.
- © PDA, Inc. 2012




{kind=link}
{kind=link}