
Abstract
Mycoplasmas are a type of bacteria that lack cell walls and are occasional cell culture contaminants. In a biotechnology setting, because they can pass through 0.2 μm filters, mycoplasmas could pose a potential patient safety hazard if undetected contaminants from the production culture were not completely removed by downstream biotechnology manufacturing. In this study we investigated the ability of typical commercial monoclonal antibody purification operations to clear and kill mycoplasmas, using Acholeplasma laidlawii as a model organism. Our spike/removal studies have shown that protein A column chromatography clears about 4–5 log10. Column regeneration effectively prevents A. laidlawii column carryover between chromatography runs. Moreover, low-pH hold steps, typically implemented after protein A purification, effectively kill A. laidlawii using either pH 3.8 glycine or acetate solutions (LRV ≥5.30 and ≥4.57, respectively). Solvent/detergent treatment, used in some processes instead of low-pH hold, also completely kills highly concentrated A. laidlawii (LRV ≥5.95).
LAY ABSTRACT: Biotechnology medicines need to be free from contaminating microorganisms such as mycoplasmas, a type of bacteria that can cause disease in humans (e.g., walking pneumonia). Here we show that some monoclonal antibody manufacturing steps can effectively clear and/or kill Acholeplasma laidlawii, a model mycoplasma species used in our study. This provides an additional level of safety assurance of biotechnology medicines for patients.
- Mycoplasma
- Acholeplasma laidlawii
- Bioprocessing
- Protein A chromatography
- Solvent/detergent
- Low-pH hold
- Spike/clearance
- Cell culture
Introduction
Mycoplasmas, of the class Mollicutes, are a type of bacteria without rigid cell walls, which allows them to efficiently penetrate 0.2 μm filters used in research laboratories and the biotechnology medicine industry. Being able to grow efficiently in primary and continuous mammalian cell cultures (1) without visible changes, mycoplasmas can establish occult infections in cell cultures (2). Thus, production of cell-derived biologics, including recombinant DNA-expressed monoclonal antibodies (mAbs) and other therapeutic protein biopharmaceuticals, can be at risk for mycoplasma contamination (3). In theory, mycoplasmas could pose a risk for patient health because some mycoplasmas can cause human disease (e.g., walking pneumonia). Therefore, regulatory guidance worldwide recommends a demonstration that biopharmaceutical products are free from mycoplasmas. Mycoplasma contaminations in facilities trigger costly, burdensome, and time-consuming facility clean-up exercises. For these reasons, mycoplasma risk mitigation steps are implemented at most biotechnology manufacturing plants (4). Such practices can include 0.1 μm filtration; heat treatment; irradiation of cell culture raw materials; and screening of cell banks, raw materials, and bioreactor cell culture harvests for potential mycoplasma contamination (5–6).
Currently, biopharmaceutical manufacturers predominantly use a culture cell– and indicator cell–based set of assays for mycoplasma detection during manufacture of cell-derived biologics, as outlined in a 1993 Points to Consider guidance document from the U.S. Food and Drug Administration (FDA) and a follow-up guidance specific to viral vaccine manufacture (7–8). The assays described are highly sensitive, but are time-consuming procedures that take up to 28 days to complete and require skilled interpretation of assay results. Nucleic acid tests (NATs) such as PCR have been advocated to replace this set of methods because of significant advantages in turnaround time (hours versus days) (9⇓⇓⇓⇓–14), and they have gained regulatory approval for this purpose on a case-dependent basis.
The detection thresholds of the two assay formats are difficult to compare, as one could imagine hypothetical examples where one would be more sensitive than the other. For example, assuming a detection limit of 50 genome copies for an NAT method, the culture method with a 1 CFU/mL sensitivity limit could be argued to be more sensitive in the case of a 100% viable, 100% monodispersed mycoplasma test article. In contrast, the same NAT test could be argued to be more sensitive in the case of a 5% viable test article where the mycoplasmas are in clumps of 200. This has led to an unresolvable debate about whether the two assay formats have comparable sensitivities, which is moot given the fact that we don't really know the phenotype of the next mycoplasma bioreactor contamination. Given this reality, the European Pharmacopoeia (Ph. Eur.) has set a sensitivity standard of 10 CFU/mL for NAT assays (15). While growth below this threshold is unlikely over long-term culture (16), some risk for NAT false negatives from very low level growth in bioreactors cannot be ruled out. Nevertheless, while NAT employed in this context might occasionally be less sensitive for detecting some mycoplasma-contaminated harvests, they have the distinct advantage of allowing for timely go/no-go decisions regarding time-sensitive in-process products.
The safety and product risk posed to biotechnology processing by using an occasionally less-sensitive method than the current culture-based set of tests in this context has not been formally studied or fully addressed. Mycoplasma survival in a vaccine process has previously been studied, but vaccine processes tend to be abbreviated compared to biotechnology processes (17). One could argue that a product risk for biopharmaceuticals would exist if mycoplasmas can survive and propagate in a biotechnology downstream process, but not if they are cleared or killed in the initial steps. Similar hypotheses are routinely evaluated for viruses in laboratory experiments through spike/removal studies using scale-down models of unit operations like columns and filters; these studies are termed viral clearance validation and routinely mitigate virus risks and support regulatory decisions for biotechnology products (18–19). The spike/removal approach has not been specifically applied to mycoplasmas in bioprocessing, but we propose that this approach can inform decision-making in the specific context of addressing the above issue regarding the appropriate sensitivity of rapid mycoplasma screening assays proposed for biotechnology drug manufacture. For example, they could begin to address the question of what happens to a completely monodispersed, low-level mycoplasma contamination if it is missed by NAT assays and enters the process stream; do they propagate? Or are they cleared or killed by the downstream processes, mitigating any product risk?
Co-culture studies with mycoplasma and typical production cells like Chinese hamster ovary (CHO) cells are very rare in the literature. While a few have been published for suspension CHO cultures (16, 20), none have used serum- or other animal component–free media typical of modern bioprocessing. Mycoplasma co-culture studies could address whether the scenario of a stealth, low-level (<10 CFU/mL) contaminant persisting for a long period in a protein-free suspension CHO cell culture is even likely.
In this report, we investigated the growth kinetics of model Acholeplasma laidlawii contamination events at high and low seeding levels in typical media formulations and raw materials utilized in commercial bioreactor cultures. We also investigated the ability of current downstream mAb bioprocessing unit operations to clear and kill mycoplasmas in spike/removal studies, using A. laidlawii as a model organism. We selected A. laidlawii as the first model organism for evaluating impacts on manufacturing because it is a potential risk for bioprocessing (21–22) and is a relatively non-fastidious species. Thus, it is a good test species for challenging standard practices in both upstream and downstream biotechnology manufacturing.
Materials and Methods
Mycoplasma Strain, Culturing, and Titering
The non-aggregating A. laidlawii strain PG8 (ATCC 23206, Manassas, VA) was used in this study (23). An ATCC strain was chosen due to the lack of availability of environmental or wild-type isolates at our site (i.e., our model bioprocessing lab doesn't have a history of mycoplasma contamination in any of our relatively limited number of bioreactor runs). A. laidlawii was cultured using SP4 glucose liquid broth and SP4 glucose agar (with thallium acetate, penicillin) (Remel, Lenexa, KS; Thermo Scientific, Waltham, MA) and incubated at 37 °C in aerobic conditions with 5% CO2. Spiking levels in the experiments ranged from 2 × 104 to 5 × 106 CFU/mL. The goal was to spike at a level where a meaningful LRV could be measured, but at the same time not so much volume that it interfered with other aspects of unit operation performance. The spike level is also limited by how high a titer is achieved during cultivation of the A. laidlawii used for spiking. Mycoplasma titers in input and output test articles were determined by performing serial 10 fold dilutions using the neutral pH, isotonic Hanks' Balanced Salt Solution (HBSS) or phosphate-buffered saline (PBS) (Gibco, Carlsbad, CA). The diluted test articles, 100 μL of each dilution, were then plated on SP4 glucose agar (with thallium acetate, penicillin). The dilution in HBSS or PBS should eliminate any residual sample matrix toxicity effects on mycoplasma on the plate. Colonies were counted 5 days after plating samples. All samples were plated in duplicate. Titered mycoplasma stock suspensions were stored at –80 °C. Stocks were thawed at room temperature before being spiked into samples. Previous studies have shown that our freeze/thaw procedures resulted in minimal loss of viability (23).
Co-cultivation of Mycoplasmas with a Suspension Culture of CHO Cells
Seed cultures of FreeStyle CHO-S cells (Invitrogen, Carlsbad, CA) were maintained at 37 °C and 8% CO2 in Thermo-Fisher CD Opti-CHO AGT Medium supplemented with GlutaMAX (Gibco). The seed cultures were initially grown in 250 mL spinner flasks (Corning Inc., Corning, NY) with a constant magnetic spinner agitation at 80 rev/min.
CHO-S cells of high viability (∼98%), as assessed using the Bio-Rad TC20 automated cell counter, were transferred from 250 cm2 flasks (Corning Inc.) into 1 L disposable spinner flasks (Corning Inc.) with CD Opti-CHO AGT medium to obtain the initial density of 1–3 × 105 cells/mL. CHO-S cells were cultured with CD Opti-CHO AGT medium supplemented with Soy Hydrolysate UF solution (SAFC Biosciences, Lenexa, KS), 1% LipoGro cholesterol supplement (RMBIO, Missoula, MT), or EX-CELL CD Hydrolysate Fusion (Sigma-Aldrich, St. Louis, MO) for 48 h or until the viable cell density reached the level of 0.5–1 × 106 cells/mL. CHO cells were then infected with A. laidlawii at the initial infection titers indicated, either ≥1 × 103 CFU/mL or ≥1 × 101 CFU/mL or left unperturbed as negative controls. Next, 1.5 mL samples were collected from the co-cultured CHO-S cells and mycoplasma on the initial A. laidlawii infection day, and then every 24 h until cell culture viability was ≤80%. Samples were stored at –80 °C until mycoplasma titering was performed.
Harvested Cell Culture Fluid for Protein A Studies and Model Protein
A murine IgG3 antibody-producing hybridoma cell line (24) was cultured in Sartorius Stedim 4 L bioreactors (Bohemia, NY) with Chemically Defined Hybridoma (CDH) media (Invitrogen) for 5 days, and then the harvested cell culture fluid (HCCF) was clarified via centrifugation (25). The resulting supernatant (HCCF) containing the model antibody was collected, 0.2 μm sterile-filtered, and stored at –20 °C until needed for experimentation.
Chromatography
Protein A mini-columns (Mab Select SuRe), with an internal diameter of 0.5 cm and bed height of 1 cm, were packed with a Tricorn 5/20 housing unit (GE Healthcare Life Sciences, Little Chalfont, UK), with coarse filter frits (porosity >25 μm) (GE Healthcare Life Sciences). All chromatography was run using an Akta Avant 25 (GE Healthcare Life Sciences) programmable system. The model capture scheme was conducted by first equilibrating the column with 10 column volumes (CVs) of PBS (100 mM sodium phosphate with 150 mM sodium chloride pH 7.2; Hoefer, Inc., San Francisco, CA). Equilibration was followed by the loading phase using the above described HCCF that was spiked immediately before the cycle was initiated with A. laidlawii (dilution factor of 103), targeting an overall process input titer of ∼105–106 CFU/mL. The resin (Mab Select SuRe, GE Healthcare Life Sciences) was loaded to an antibody dynamic binding capacity (DBC) typical of commercial manufacturing (80%) based on vendor-claimed DBC. The salt wash phase was operated using 30 CV of one of two solutions, PBS (100 mM sodium phosphate with 150 mM sodium chloride pH 7.2) or 100 mM sodium phosphate with 1 M sodium chloride pH 7.2. The elution phase used 25 CV of one of two elution solutions that differed only in their pH (100 mM glycine, pH 3.5 and pH 4). Between 2 and 12 μL of 3 M tris pH 8.6 were pre-loaded into tubes used for elution fraction collection in order to rapidly bring the elution fraction to neutral pH. 0.1 M NaOH (50 CV) was used as a column sanitization step in between each run. Loading phase, flow-through, salt wash, and elution fraction test samples were collected, titered as described above, and then volume-adjusted to obtain a total A. laidlawii count in each fraction.
Statistical Analysis and Design
Analysis of the downstream processing data from the full factorial design of experiment (DoE) of chromatography was performed in JMP (Ver. 11.1.1, SAS Institute Inc., Cary, NC). Logarithmic reduction values (LRV) were calculated from the loading phase flow-through, salt wash, and elution fraction volume-adjusted mycoplasma counts. For experiments that resulted in no visible colonies on the solid agar medium, the presumed limit of detection (LOD) of the culture-based assay (10 CFU/mL) was used for LRV calculations. A cutoff of P < 0.05 was used to select significant factors to be evaluated in follow-up column carryover experiments. The LRVs for solvent/detergent (S/D) treatment and low-pH hold experimental time points were expressed in the accompanying figures as mean ± standard deviation. For the cell culture studies, analysis of variance (ANOVA) analyses were run with a cutoff of P < 0.05 for significance.
Column Carryover Experiments
To test for column carryover, each experimental replicate was run as a two-cycle purification scheme. The first cycle (Cycle One) was run according to the best, worst, and center point parameters for flow rate, wash solution salt concentration, elution solution pH, and regeneration detailed in Figure 5A. If the experimental design called for a regeneration step, the column was washed with 30 CV of 6 M urea (for worst-case and center point conditions) and then re-equilibrated with PBS (30 CV). Once Cycle One was completed, four additional steps were carried out; these will be referred to as Cycle Two. Cycle Two was conducted by first taking the column off-line (i.e., column position changed to bypass). The system, which included the sample pump and lines, was sanitized with 0.1 M NaOH (25 mL) and pH quenched with equilibration solution (25 mL). The column was then loaded through the sample pump with equilibration solution, and the effluent (6 mL, fixed volume) was collected as the blank run for mycoplasma titering. Equilibration phase effluents were chosen for measurement as a worst case for potential carryover (i.e., immediately after the first cycle). As this sampling point has no set volume, carryover data is expressed as CFU/mL in the effluent, rather than an LRV. Once the blank run was collected, the column was sanitized with 30 CV of 0.1 M NaOH and quenched with 30 CV of equilibration solution. Cycle One load phase, salt wash, elution, and Cycle Two blank run fractions were collected for mycoplasma titering as described above.
Low-pH Hold Experiments
A model IgG3 antibody solution was obtained by running the above described HCCF over a protein A column on an Akta Avant 25 system using the solutions described above. Samples were eluted using one of two elution solutions: 100 mM glycine pH 3.5 or 100 mM acetate pH 3.2. Collected elution fractions were pH-adjusted to pH 3.8 using 2 M acetic acid and 0.5 M tris base titrants, respectively, and then spiked with A. laidlawii (t = 0 min, load control) at a dilution factor of 103 (5 μL of A. laidlawii stock suspension into 5 mL of eluate) for an overall sample titer of ∼105–106 CFU/mL. Post-spike sample pH was verified to not significantly alter following the addition of A. laidlawii spike. The A. laidlawii–spiked eluates were incubated for 60 min at 25 °C and sampled at 0, 5, and 60 min for titering as described above. Other column solutions were evaluated for mycoplasma killing using similar procedures (Figure 3).
Solvent/Detergent (S/D) Treatment
HCCF was spiked with A. laidlawii as described in the Low-pH Hold Experiments subsection above (1:1000 ratio of A. laidlawii stock suspension to HCCF). Tri(n-butyl)phosphate (TNBP) as solvent and Tween 80 as detergent were added and mixed into the A. laidlawii-spiked HCCF in one of two proportions (0.1% TNBP/0.5% Tween 80 and 0.001% TNBP/0.005% Tween 80) (26). Industry standard conditions of 0.1% TNBP/0.5% Tween 80 represent the lower boundary of S/D proportions used in commercial settings. Failure point conditions of 0.001% TNBP/0.005% Tween 80 is 100 fold more dilute. A. laidlawii load control samples were taken and titered before adding S/D (t = 0 min). S/D-treated test articles were incubated at 25 °C and sampled at 5 min and 60 min after the addition of S/D. Samples were then titered as described above.
Results
To evaluate mycoplasma growth and survival in model bioprocess unit operations, A. laidlawii titers in process inputs and outputs were determined by colony counting using a 5 day culture method. While all culture assays experience some variation, our results remained within the same order of magnitude between experimental replicates. Test articles that resulted in no plate colonies were considered below the assay LOD (10 CFU/mL).
Growth of Mycoplasma in Suspension CHO Culture
Spinner cultures of CHO cells were spiked at either high or low levels (or not, as a control) with A. laidlawii (Figure 1). The first level, ≥103 CFU/mL, was a positive control condition with a spiking level sufficiently dense to favor the contaminant taking hold in various culture conditions. A second spiking level, ≥101 CFU/mL, was done to mimic a culture with a live mycoplasma contaminant that may evade detection by an assay meeting the Ph. Eur. LOD requirement of 10 CFU/mL. As can be seen (Figure 1A), under 2 of 4 conditions, addition of 103 CFU/mL mycoplasma had a sufficient seeding density to take hold. The exceptions were Opti-CHO without any supplementation, and Opti-CHO supplemented with LipoGro; neither appeared to support A. laidlawii growth, as no detectable levels were found after 24 h. In contrast, A. laidlawii grown in cultures containing hydrolysates expanded 100–1000 fold after 24 h and persisted at high levels in culture for at least 3 days. When spiked at a low level in the presence of hydrolysates (Figure 1B), the A. laidlawii grew to >107 CFU/mL. This was a larger increase in detectable A. laidlawii titer than was observed with the higher spike level in Figure 1A, suggesting that persistent low-level contaminations of regulatory concern would more likely expand up and be detectable within 24 h. For both spike levels, A. laidlawii persisted at measurable levels for up to 96 h, although some supplemented culture conditions begin to see waning of A. laidlawii growth after 72 h.

Growth kinetics of A. laidlawii known contamination levels in various CHO-S spinner cell culture media. (A) High-level contamination spike kinetics bar graphs represent A. laidlawii CFU/mL obtained from each 24 h sample with time 0 as day of initial spike. CHO-S spinner cultures were targeted to be spiked with ≥1 × 103 CFU/mL A. laidlawii spike into 72 h CHO-S spinner cultures (modest T = 0 variation represents volume and titration error). “—” represent cell densities taken at sample times. (B) Bar graphs represent A. laidlawii CFU/mL obtained from each 24 h sample with time 0 as day of initial spike. CHO-S spinner cultures were targeted to be spiked with ≥1 × 101 CFU/mL A. laidlawii spike into 72 h CHO-S spinner cultures. “—” represent cell densities taken at sample times. (C) CHO cell growth curves of cultures where initial mycoplasma spike did versus did not measurably grow. Combined data encompasses both spike levels and all tested media conditions of cultures represented in Figure 1A and 1B.
To analyze the impact of the presence of A. laidlawii on the CHO cells in culture, the kinetics of CHO cell viable cell densities were compared between all cultures in which the A. laidlawii contamination took hold and those that did not (i.e., cultures where no detectable levels of A. laidlawii were observed after 24 h). In CHO cultures where the A. laidlawii contamination took hold, only subtle differences in viable cell density and growth rate were apparent (Figure 1C), arguing that general decline in the overall growth rate of a bioreactor culture is not a very robust indicator for potential A. laidlawii contamination events. Although we were not able to monitor other potential culture indicators in our spinner system (e.g., dissolved oxygen, CO2), it may be that there is no single “perfect indicator” of when an occult A. laidlawii contamination occurs.
A. laidlawii Fractionation through a Protein A Capture Column
Protein A bind and elute chromatography has six or more distinct stages (Figure 2A): equilibration, column load with HCCF, post-load wash with equilibration solution, secondary wash with high salt or other more stringent solutions to remove non-specifically bound proteins, post-secondary wash equilibration, elution of the antibody by a low-pH solution, and regeneration using a chaotropic or other solution to remove residuals present on the column. A scaled-down model capture process was designed to have the above six stages for mycoplasma spike/removal studies using a non-clumping form of A. laidlawii. A non-clumping strain was chosen to minimize artificial overestimation of the column clearance capacity by trapping of clumped mycoplasma in the column frits. Protein A mini-columns were used in the model capture chromatography because mini-columns would have a shorter contact time compared to commercial chromatography, representing a worst-case condition for chromatographic resolution. The mini-column purification scheme also allowed for rapid elution, which minimized the amount of A. laidlawii killed in the column by the low-pH front during elution before the collected eluate pool could be quenched and titered. This was an important factor considering our A. laidlawii quantification method was based on viable CFUs.
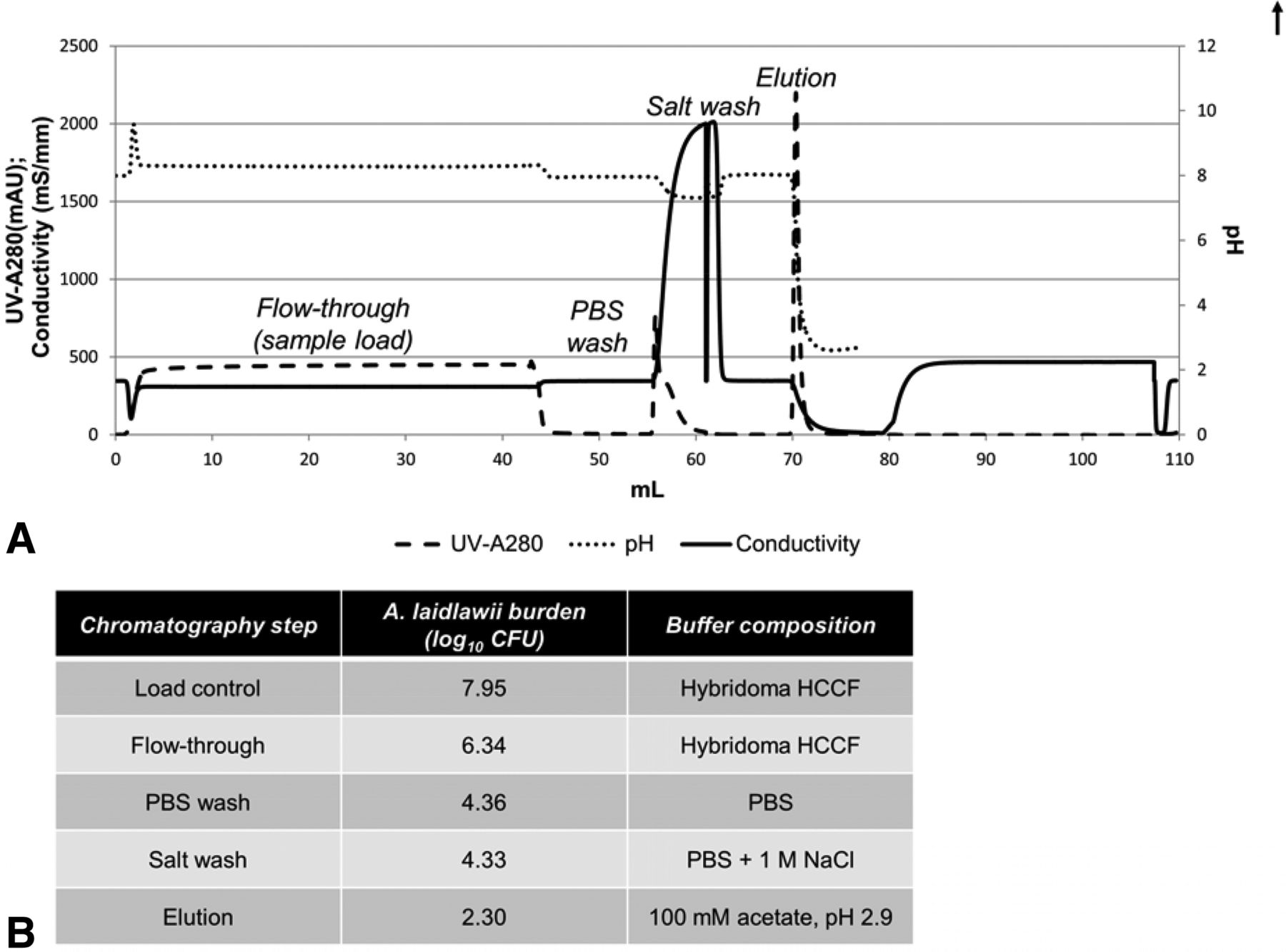
Partitioning of A. laidlawii during a typical protein A purification step. (A) A typical protein A purification chromatogram, with peaks labeled with the corresponding fraction collected. The pH probe was taken off-line following elution to protect it during column regeneration and sanitization steps. (B) Total A. laidlawii CFU present in fractions collected during protein A purification of A. laidlawii-spiked HCCF. The elution fraction was in-line neutralized using 3 M tris (pH 8.6).
Because of the bind and elute nature of protein A chromatography, it is likely that the majority of mycoplasma would flow through the column during the load; residuals non-specifically binding could be further cleared by a salt wash. Our experimental observations bore out this hypothesis; most A. laidlawii burden is present in the flow-through after loading, with only low levels persisting until the elution phase (Figure 2B). This results in an LRV of 5.7 log10 CFU for this particular protein A purification process.
A. laidlawii Survival in Protein A Chromatography Solutions
In order to accurately understand the partitioning of A. laidlawii in a protein A column, it is useful to understand the potential confounding factor of A. laidlawii inactivation by the chromatography buffers. To illuminate this, A. laidlawii was spiked directly into a variety of solutions typically used in protein A chromatography for equilibration, secondary wash, elution, regeneration, or sanitization (Figure 3). Of the secondary wash solutions tested, only citrate demonstrated moderate (1.13 log10) inactivation of A. laidlawii after a period of 15 min; neither PBS nor high NaCl salt solution significantly inactivated A. laidlawii, even after an incubation period of an hour (Figure 3A). By contrast, the column regeneration and sanitization solutions completely killed A. laidlawii by 15 min post-spike (Figure 3C). This was expected because the solutions that typically used for column regeneration and sanitization are very strong bases or chaotropic agents. As for elution solutions, most of the moderately acidic buffers tested inactivated A. laidlawii by 15 min of exposure (Figure 3B). Only in glycine solutions, a weakly acidic amino acid solution when at a relatively high pH of ≥4.0, did a residual amount of A. laidlawii survive to 15 min post-spike.
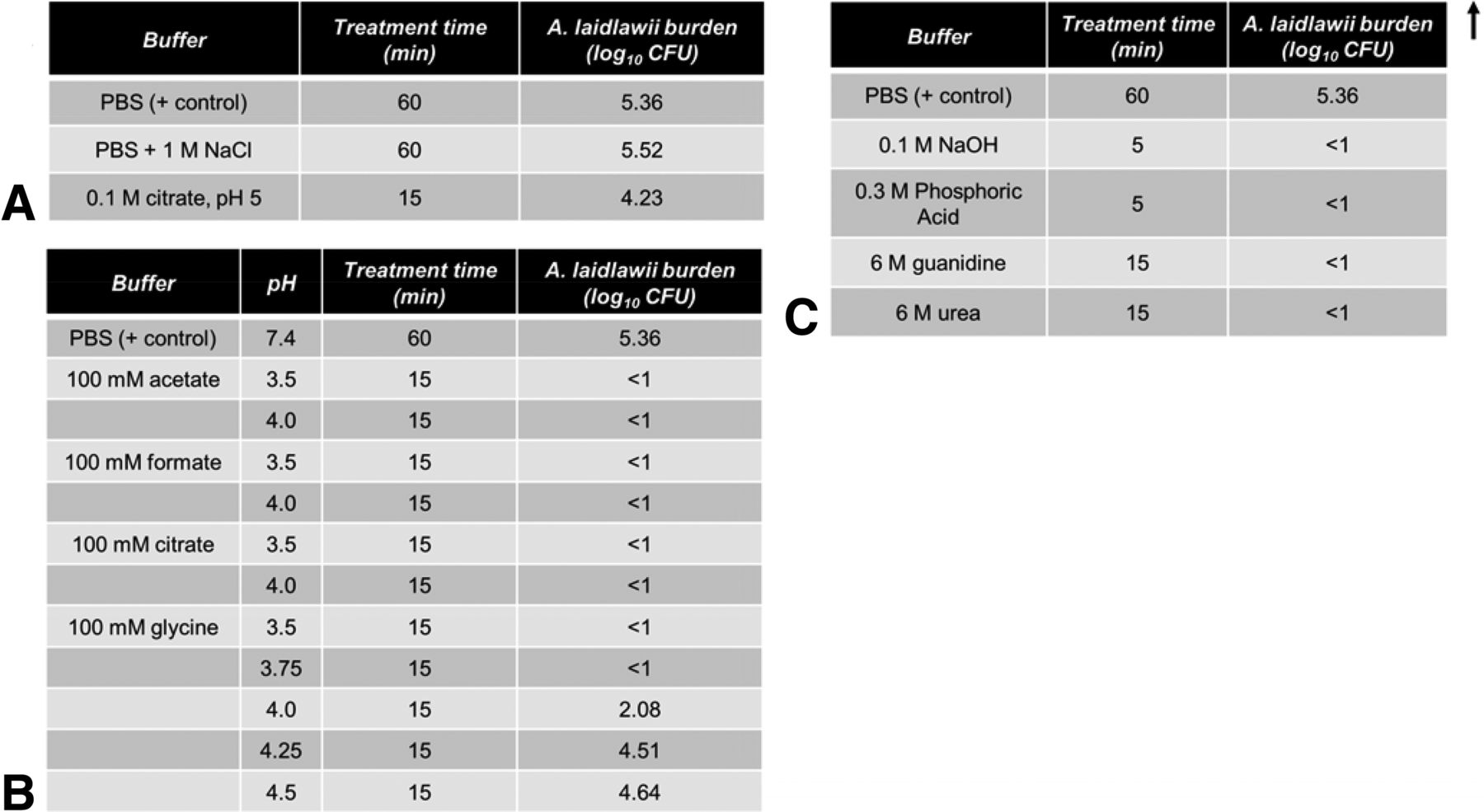
Viability of A. laidlawii following exposure to solutions commonly used in protein A affinity chromatography. A. laidlawii was spiked into each solution, mixed thoroughly, and incubated at 25 °C. Samples were taken at various time points and titered. The average mycoplasma load was 2.30 × 105 CFU/mL (1.96 × 106 total CFU, volume-adjusted for reaction volume). (A) Common post-loading wash solutions. (B) Common elution solutions. (C) Common sanitization and column regeneration solutions.
These results informed our DoE study of protein A chromatography (see below) in terms of the worst-case conditions. As worst case would be conditions where buffer inactivation of mycoplasma is minimized, we selected the low/high–salt solutions for wash solutions (PBS and PBS + 1 M NaCl, respectively), the 100 mM glycine solution for elution (low/high pH of 3.5 and 4.0, respectively), and 0.1 M NaOH as the column sanitization solution.
Protein A Design of Experiment (DoE)
To confirm this observation and to determine factors that may affect mycoplasma clearance, a full-factorial DoE was conducted. In selecting the chromatography parameters, a worst-case scenario theme was applied such that the solutions chosen reflected parameter settings that could be argued to be at process extremes with lower mycoplasma clearance capacity. A full factorial DoE was used to initially evaluate the effects of three parameters (system flow rate, wash solution salt concentration, and elution solution pH) chosen based on plausibility of impact (Figure 4B). These parameters were identified based on scientific principles predicting their ability to affect the potential interactions of the mycoplasma with the protein A resin or mycoplasma viability (27). For example, system flow rate would affect the residence time, which, in theory, could affect the mycoplasma's ability to interact with and immobilize, or get trapped in, pores on the column. The flow rate could also theoretically change the contact time of the low-pH elution with any mycoplasmas bound non-specifically or trapped in pores. The wash solution salt concentrations modeled a secondary wash (stringent versus weak) that clears non-specifically bound impurities, which is a typical step in protein A purification. Finally, as more acidic solution systems are predicted to negatively affect mycoplasma viability, the elution solution system and pH were selected based on the upper pH range likely in commercial manufacture. For each set of run parameter patterns, the Akta Avant 25 was programmed to purify a mycoplasma-spiked HCCF sample load, sending effluent flow into a fraction collector during the load phase, salt wash, and elution.

Factors affecting clearance of A. laidlawii by protein A affinity chromatography. The average HCCF mycoplasma load was 5.47 × 105 CFU/mL (3.03 × 107 total CFU, volume-adjusted for average milliliters of HCCF loaded). (A) Three chromatography parameters were each tested at two levels. (B) A. laidlawii burden in the flowthrough and salt wash fractions collected from each of the 16 patterns. LRV calculated from the A. laidlawii burden in the elution fractions. (C) Standard least squares analysis of parameters affecting A. laidlawii titer in the salt wash fraction. (D) Standard least squares analysis of parameters affecting A. laidlawii titer in the elution fraction. (E) Existing interaction effects between parameters affecting A. laidlawii titer in the elution fraction.
Precautions were also taken to specifically evaluate mycoplasma removal vs. mycoplasma kill by low pH elution solutions. For example, because of the small column size and relatively large dead volume present in the lines, large CVs were used to ensure that there was a clear separation between chromatographic steps. Immediate pH neutralization of eluates was done to maximize A. laidlawii survival in the elution fractions, thus minimizing potential confounding effect of them being killed first by the acidic pH of the elution solution.
As can be seen, the LRVs of the 16 different experimental runs in the DoE varied between 3.1 to 5.4 log10 (run 6 and 3, respectively; Figure 4B). Salt wash molarity was the prime factor that correlated in a consistent manner with LRV (Figure 4C). Although elution solution pH did not have such a clear correlation, the P-value of 0.06 indicated that it may be important (Figure 4D). Therefore, elution pH was included for further study in our follow-up experiments on mycoplasma carryover between column runs. The interactions between the tested parameters were not observed to have a significant effect on overall process LRV (Figure 4E).
Mycoplasma Persistence between Chromatography Cycles
The above experiments indicated that not all A. laidlawii are removed/killed immediately after undergoing a single protein A purification cycle. To determine if A. laidlawii can survive an initial cycle and be carried over to subsequent cycles, each of three sets of chromatography parameter patterns was run in a two-cycle design; the second cycle of each chromatography run was sampled to test for A. laidlawii presence in the second cycle effluent (Figure 5). Based on the DoE results, three parameter patterns were selected (best-case DoE pattern #15, worst-case DoE pattern #6, and center point) and run in triplicate (Figure 4B–E). The center point run designed between the two extremes (Figure 5A) provided a measure of process stability and inherent variability. In addition, a column the regeneration step was only included in the best-case purification scheme. We chose 6 M urea as the regeneration solution because it is a common commercial protein A cleaning solution but not as harsh as other sanitization steps for microbial kill (e.g., NaOH) (28–29).
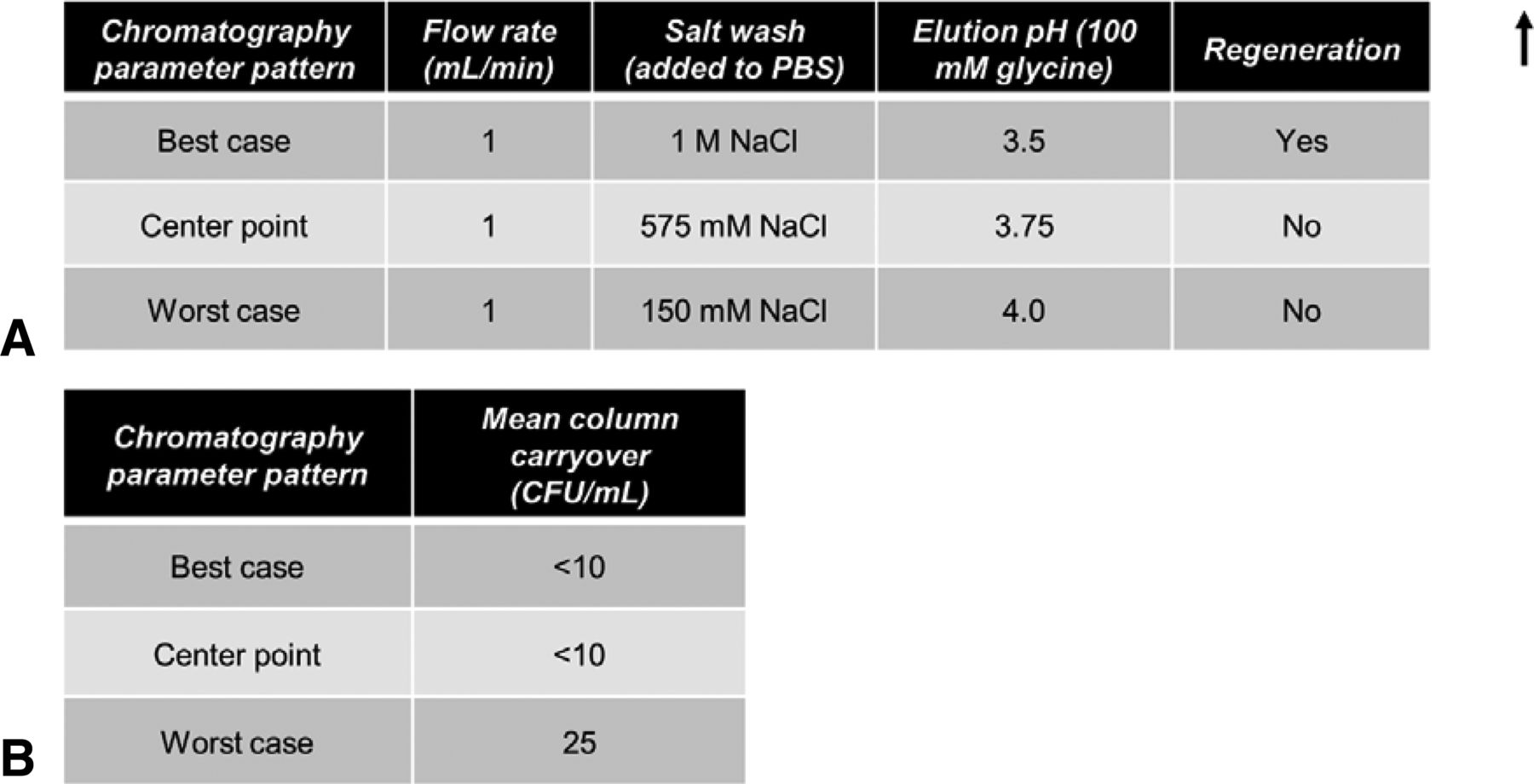
Evaluation of potential for A. laidlawii column carryover. In the two-cycle purification scheme, the average HCCF A. laidlawii load for the first cycle was 2.99 × 105 CFU/mL (9.36 × 108 total CFU, volume-adjusted for average mL of HCCF loaded). No additional A. laidlawii was spiked into the second cycle. (A) Chromatography parameters for the three chosen conditions. Elution solution, 100 mM glycine. Regeneration was 30 column volumes of 6 M urea. Residence time, 11.76 s. (B) Column carryover observed for best-case, worst-case, and center point chromatography conditions.
As can be seen in Figure 5, only in the worst-case scenarios were mycoplasmas detected in the two-cycle effluent, and only 25 CFU/mL were present. Thus, while under the “no regeneration” condition some mycoplasmas can survive, they did not seem to multiply. In a separate study (Figure 3C), four common column cleaning solutions were tested for A. laidlawii killing ability in solution phase. In each case, 5–15 min of contact time resulted in complete kill of 105 CFU/mL of A. laidlawii, with an LRV of ≥4.4 log10. Two common secondary wash solutions were also tested for A. laidlawii killing ability (Figure 3A). In contrast to the column cleaning solutions, the secondary wash solutions did not significantly decrease the number of viable A. laidlawii CFU even after 60 min of contact time.
Effect of Low-pH Hold on Mycoplasma Viability
In these studies we examined the kill kinetics of A. laidlawii spiked in two solution species used for protein A elution and subsequent lipid-enveloped virus inactivation holds, glycine and acetate at pH 3.8 (Figure 6). According to available published data, this pH level, pH 3.8, represents a worst-case condition for lipid-enveloped virus inactivation (27, 30). In both cases, a significant decline in viability (approximately 3 log10) was evident by 5 min, and complete kill (LRV ≥4.6–5.3 log10) was achieved by 60 min. A 60 min timeframe is at the lower limit of typical low-pH incubation steps in industrial settings. To determine the pH sensitivity of the inactivation afforded by low pH, glycine solution with a range of pH levels (3.5 to 4.5 in 0.25 unit increments) were evaluated for mycoplasma kill efficiency (Figure 3B). All solutions at pH 3.75 or less killed concentrated A. laidlawii (105 CFU/mL) after 15 min, while at pH 4.0 a low level residual persisted. Solution at pH 4.25 was completely ineffective.
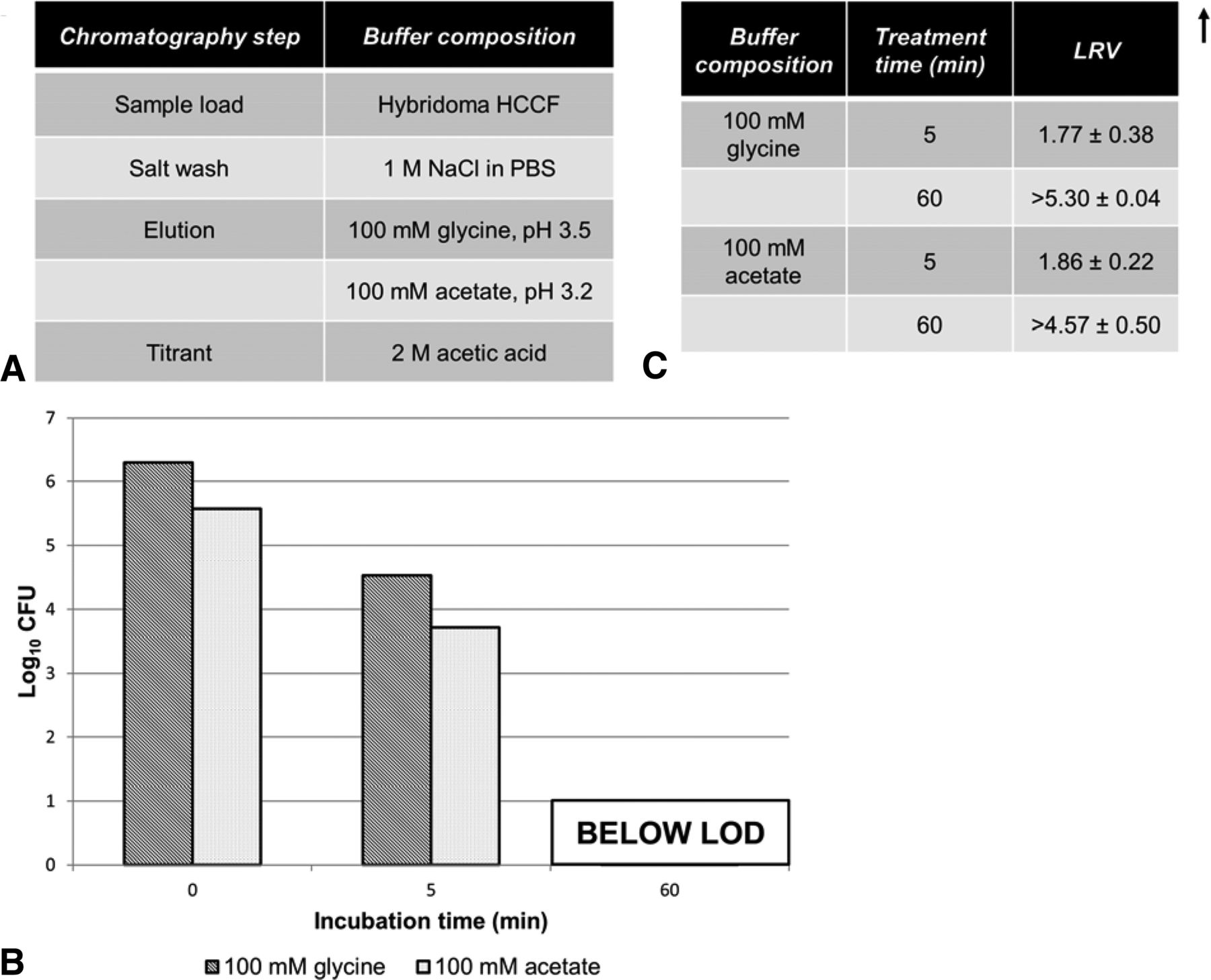
Viability of A. laidlawii after low-pH hold. Antibody-containing eluate was titrated to pH 3.8, then spiked with A. laidlawii and incubated for 60 min. The average elution fraction mycoplasma load was 2.51 × 105 CFU/mL (1.25 × 106 total CFU, volume-adjusted for mL of elution fraction). (A) Solutions used during protein A purification for low-pH hold experiments. (B) Samples were taken at time 0 min (load control), 5 min, and 60 min, and titered. (C) LRVs calculated from A. laidlawii titers at 5 min and 60 min time points.
Solvent/Detergent (S/D)
We treated A. laidlawii for 5–60 min with S/D in a procedure specified by the New York Blood Center for inactivating lipid enveloped viruses (31). As a negative control, the same A. laidlawii preparation was treated with a 100 fold more dilute S/D solution than what is currently used in industry, which we identified as the failure point for this treatment (Figure 7). As can be seen, the standard S/D procedure rapidly killed the A. laidlawii, with an LRV ≥6.0 log10 by 60 min.
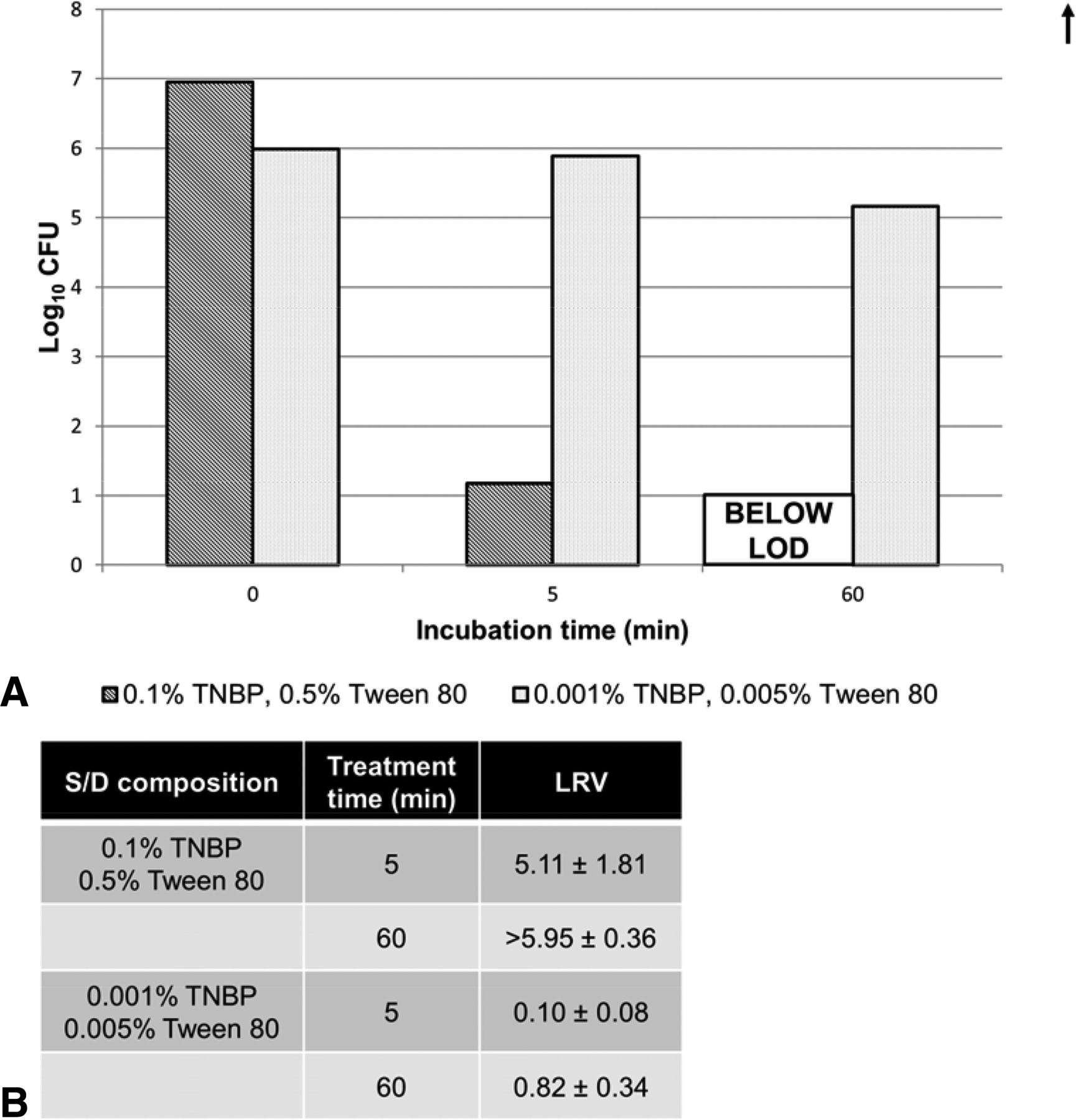
Viability of A. laidlawii after solvent/detergent (S/D) treatment. A. laidlawii–spiked HCCF was treated with different amounts of S/D. The average HCCF mycoplasma load was 1.59 × 106 CFU/mL (6.38 × 106 total CFU, volume-adjusted for milliliters of HCCF test sample). (A) Samples were taken at time 0 min (load control before addition of S/D), 5 min, and 60 min, and titered. (B) LRVs calculated from A. laidlawii titers at 5 min and 60 min time points.
Discussion
Mycoplasmas have the potential to grow and contaminate bioreactor-based mammalian cell cultures. For this reason, bioreactor harvests are tested for mycoplasma as part of the overall biotechnology product quality and safety assurance strategy (13). Currently, mycoplasma testing relies mostly on culture methods developed in the 1970s and 1980s, promulgated in a 1993 document from the Center for Biologics Evaluation and Research/FDA (7–8). These tests, referred to colloquially as the Points to Consider tests, can test large volumes of sample and are assumed to be capable of amplifying and cultivating any live mycoplasma in a test article. These tests, however, can take up to 28 days to complete, a timeframe inconsistent with rapid bioprocessing. Approval actions of nucleic acid tests as rapid alternatives for the Points to Consider method have taken place on a case-specific basis, but the worldwide approval pathway in general is an evolving landscape, and it could benefit from a realistic assessment of the actual risk posed by mycoplasma to the entire process. Exact equivalence between test results and LOD has proved elusive because they measure different aspects of the test article, genome copies (GC) versus CFU. These two attributes do not correlate on a one-to-one basis, as would be expected in a 100% viable, monodispersed test article. One clump of 1–200 mycoplasma would grow as one colony on plated agar media, yet would contain 100–200 GC. A 10% viable culture would have 10 fold higher signal in a NAT test, which would measure the GC in all mycoplasma, including the non-viable cells. The degree of clumping and mycoplasma viability can vary with the culture conditions used to prepare a test article or reference standard (16, 23, 32⇓–34).
With all this complexity in mind, the Ph. Eur. has proposed 10 CFU/mL as a workable LOD for NAT assays when tested in a panel of mycoplasma species. The NAT assay would need to be demonstrated to be robust and insensitive to the various components in matrix material (i.e., proteins, extraneous nucleic acids, etc. in the extracted or otherwise processed harvest material). From a regulatory standpoint, the theoretical risk associated with harvest testing using methods designed to meet this standard would be the potential for very low–level contaminants to be missed by the harvest screen and be forward-processed into downstream purification. There are two elements that contribute to this risk: (1) How likely is a very low level contaminant to persist at that level in culture (i.e., below the Ph. Eur. LOD of 10 CFU/mL) without dying off? (2) Would mycoplasma survive once they enter the purification stream?
To address question 1, we performed CHO-S and A. laidlawii co-culture experiments to track mycoplasma persistence and growth. In general, A. laidlawii will grow several fold within 24 h in co-culture if supplied with key elements present in hydrolysate supplements, but these seem to be limiting by 96 h. Our results were consistent with previous findings from the co-culture of two mycoplasma species in CHO-S cells in serum-containing media (20). In summary, it seems that if an A. laidlawii contamination takes hold, it will grow and stay within measurable levels by a test that complies with the Eur. Ph. sensitivity standard. If it does not take hold, it is probably because chemically defined media like Opti-CHO are missing an unidentified essential chemical nutrient that appears to be present in hydrolysates and fetal bovine serum. However, persistent low-level contaminants do not seem to represent a risk factor for NAT assays in that, as seen in Figure 1A and 1B, the co-cultured mycoplasma do not persist long as a stealth contaminant and are instead well above the Ph. Eur. LOD of 10 CFU/mL within 24 h of cell culture.
To address question 2, we performed spike/clearance studies of three common bioprocessing unit operations using model process intermediates spiked with A. laidlawii: protein A chromatography, low pH hold, and S/D treatment. The first unit operation, protein A chromatography, is the most common capture step for mAb processes (Figure 2). While most of the spiked A. laidlawii flowed through the column unbound during the loading phase, some residuals co-eluted with the product. Our DoE experiments revealed that salt wash molarity was the only factor that correlated in a consistent manner with LRV. This correlation, higher LRV with a higher molarity salt wash is consistent with the biology/engineering of the step in that the more stringent salt wash would strip additional residuals from the column over those that merely flowed through.
Nevertheless, the low levels of A. laidlawii in the eluate pool indicated that not all mycoplasmas were cleared immediately or flow-through unbound during a protein A column unit operation. In theory, residual microorganisms left on a column after a purification cycle could multiply if sufficient nutritive factors exist on the column. Our above observation that it is difficult to grow mycoplasma even in highly nutritive Opti-CHO media argues that this is unlikely. In practice, column sanitization solutions are designed to kill any microbial contaminant to prevent microbial growth. This step is typically conducted either every cycle or at most after five cycles, which can be required to process a harvested bioreactor. Our solution hold studies (Figure 3C) demonstrated that pH extreme solutions, 0.1 M NaOH and 0.3 M phosphoric acid, are extremely efficient at killing A. laidlawii, indicating that a protein A column sanitized with one of those solutions would not allow A. laidlawii to survive prior to resin storage between campaigns. Depending on the resin manufacturer and resin chemistry, high molarity chaotropic agent regeneration solutions can be chosen instead. Our model solution, 6 M urea, kills A. laidlawii in solution phase and in column carry-over studies. Only at worst-case conditions without a salt wash and/or any cleaning/regeneration cycle did a residual of A. laidlawii persist on the column.
Low-pH inactivation is a common step in antibody bioprocessing that is typically introduced following the protein A capture step to inactivate retrovirus-like particles (RVLPs) produced by the expression CHO or myeloma cells. The capture eluate intermediate is typically held 1–2 h at room temperature with the pH adjusted to 3.6 or less. This step is so robust for RVLPs and other lipid-enveloped viruses that an ASTM standard was promulgated specifying conditions where 5 log10 clearance could be expected for rodent retroviruses like RVLPs (35). The inactivation of ≥4.6–5.3 log10 A. laidlawii by 60 min supports our hypothesis that conditions employed to inactivate RVLPs would also kill A. laidlawii. Mycoplasmas are bacteria that lack a cell wall but possess a lipid bilayer cell membrane, and thus they are likely able to be killed by steps designed to inactivate lipid-enveloped virus like RVLPs (1). Although the low-pH hold was less effective at pH 4.25 (Figure 3B), this step is rarely performed at pH 4.0 or above in industrial practice because RVLP inactivation is also highly sensitive to pH. Furthermore, ASTM standard E2888-12 specifies a pH of 3.6 or less for this step; thus, if a manufacturer follows the ASTM standard, any A. laidlawii would be killed along with the RVLPs.
S/D treatment is a procedure originally designed in the 1990s by the New York Blood Center to inactivate retroviruses like HIV in blood and plasma products (31). While used extensively in the plasma product industry (19), it is also employed by some commercial antibody processes in lieu of low-pH inactivation, particularly for antibodies that may be pH-sensitive. As this step is designed to inactivate lipid-enveloped viruses (i.e., HIV) by dissolving their lipid membranes, it also may kill the cell wall–deficient mycoplasma. The rapid inactivation of A. laidlawii by S/D treatment within 60 min provides assurance for safe processing following this step, as it is at the lower end of incubation times for this step in commercial practice.
In summary, we found that A. laidlawii is unlikely to persist for long at a low-level contamination below the Ph. Eur. LOD in a bioreactor cell culture, especially in a commercial environment where a set number of days would exist between the last bioreactor feed and harvesting. Furthermore, steps currently implemented by biotechnology firms to capture the antibody or inactivate RVLPs, like low-pH inactivation or S/D treatment, remove orders of magnitude or achieve complete kill of high-titer preparations of A. laidlawii. We contend that our results can be generalized to processes that employ these steps in a product-independent manner, and we do not advocate the need for process validation-like mycoplasma spike/removal studies on a product-by-product basis. It could be argued that other mycoplasma species or wild-type strains are more resistant to such factors as low pH because of protective effects of aggregation. However, the fact that all mycoplasma lack a cell wall (making them more vulnerable to inactivation by chemicals than other bacteria) and that we chose a relatively non-fastidious type of mycoplasma argues that our results with A. laidlawii are a reasonable first model of how mycoplasmas would react and survive in a bioprocessing environment. While in theory repeated exposure of wild-type mycoplasmas to bioprocessing environments could exert evolutionary pressure towards resistance, firms tend to inactivate and discard contaminated bioreactor cultures, stopping the evolution process at that point. Further, our A. laidlawii growth kinetics data indicate that low-level contaminant would be unlikely to persist below a detectable level by a Ph. Eur. compliant NAT-based assay for very long. Nevertheless, we intend to follow up these data with an evaluation of a panel of other, evolutionarily and metabolically distinct mycoplasma species. Overall, our results to date support the contention that mycoplasma test methods complying with the Ph. Eur. LOD standards of 10 CFU/mL possess suitable sensitivity to assure product safety in the context of typical mAb bioprocessing.
Conflict of Interest Declaration
The authors declare that they have no competing interests.
Disclaimer
The opinions discussed in this paper are those of the authors and do not necessarily reflect official FDA views or policy.
Acknowledgements
We acknowledge the Center for Drug Evaluation and Research (CDER) Regulatory Research Coordinating Committee for funding this project. This project was supported in part by an appointment to the Research Participation Program at FDA/CDER/OBP administered by the Oak Ridge Institute for Science and Education through an inter-agency agreement between the Department of Education and the FDA. We acknowledge Dr. David Frucht and Laurie Graham (CDER/FDA) for insights and ideas into how to address the issue of mycoplasma testing and fate in manufacturing. We acknowledge Alexandra Markus from the University at Buffalo for her assistance with experiments.
- © PDA, Inc. 2017




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}