
Abstract
Container–content compatibility studies are required as part of the submission of a new product market authorization file or for a change relating to the primary product-contact packaging. Many regulatory publications and guidances are available in the USA, Europe, and Japan. However these publications and guidances are not sufficiently precise enough to allow for consistent interpretation and implementation of the technical requirements.
A working group has been formed by the French Society of Pharmaceutical Science and Technology (SFSTP) in order to propose guidance for container–content interaction studies that meet both European and US requirements, and allows consistent and standardized information to be presented by the industry to the regulators.
When a pharmaceutical drug product remains in prolonged contact with a material, the two critical points to consider are the drug product's quality and safety. A pharmaceutical evaluation of the container–content relationship should be done based on the knowledge of the contact material (e.g., type, physicochemical properties), its manufacturing processes (e.g., the type of sterilization that could potentially alter the interactions), and the formulation components involved in contact with this material (e.g., physicochemical properties, pharmaceutical presentation, route of administration). Quality is evaluated using the stability study performed on the product. Safety is partially evaluated with the stability study and is analyzed in conjunction with toxicity testing, specifically with cytotoxicity testing. The toxicity aspect is the key point of the container–content compatibility study and of patient safety.
Migration tests are conducted when an interaction is suspected, or found based on previous results, to identify the component responsible for this interaction and to help select a new material if needed. Therefore, such tests are perhaps not the best ones to use for the purpose of safety evaluation. Consequently, a decision tree based mainly on the toxicity aspect is proposed in order to support the pharmaceutical companies' container–content interaction approach and filing.
Introduction
All pharmaceutical products are in contact with a construction material of one sort or another during the manufacturing process or storage up to the point of use. This material can interact and modify the quality of the contents and/or the material itself. The potential interaction is determined by the material type, the physicochemical properties of the contents, and the pharmaceutical formulation. Plastics and elastomers are considered to have the highest potential interaction with the content. However, other materials often considered to be inert, such as glass and stainless steel, cannot be neglected. Moreover, the route of administration must be considered in order to define the consequence of these interactions and, if appropriate, to launch specific studies.
Compatibility studies must be included in the dossier submitted for a marketing authorization whenever a new product is introduced or when an after post-approval variation related to a material change occurs. Several pharmaceutical and food regulatory publications are available in Europe and in the United States of America (US). Currently, there is no specific and accurate regulation from the International Conference on Harmonization (ICH) applying to the three regions (Europe, North America, and Japan). Some regulatory requirements for food products can be found in Europe and in the US. All these regulatory requirements focus on the quality of the material, in particular the potential threat to safety. These requirements apply to the different steps of the life cycle of a pharmaceutical product: from the development or the selection of a material in contact with the product up to and including any post-approval change to assess the material's compliance with the pharmaceutical product. For this assessment, the product manufacturer has to
-
get the complete information related to the material for the intended use
-
define the approach, based on this information availability and the product characteristics
-
have a very strict and objective interpretation of the results, assessing the impact on the efficacy and the safety of the product
The selected approach must not lead to overestimating the material impact on the safe use of the product, and must be included in a wider analysis of the quality of the product and the material. The methodology must be based on a scientific approach and must clearly define the container–content compatibility study's objective.
Regulatory Publications
Several regulatory publications are available regarding pharmaceutics and food in Europe, including The European Medicines Agency's EMEA Guidelines (1) and the European Pharmacopoeia (Ph. Eur.) (2). The US provides the Guidance for Industry (3, 4), the Code of Federal Regulations (CFR) (5–7), and the United States Pharmacopoeia (USP) (8). Japan provides the Japanese Pharmacopoeia (JP) (9), and at the international level there are the standards of the International Organization for Standardization (ISO), a network of the national standards institutes of 161 countries (10).
The EMEA Guideline on Plastic Immediate Packaging Materials (CPMP/QWP/4359/03) (1), introduces the new dossier format. In particular it introduces the quality aspect in which information on the primary packaging has to be provided, and it relates to plastic materials used for active ingredients and final product packaging, except for elastomers. Decision trees, based on the drug product's physical form and administration route, are provided, as well as the material compliance to a pharmacopoeia. The requirements are presented in terms of general information, specifications, and, extraction and migration studies. Migration studies are not required for solid products used for oral and topical application (not including ophthalmic use). Note that migration studies can be considered for solid products intended for parenteral or ophthalmic use or for inhalation. In all other cases migration studies are required and a toxicological analysis is to be conducted if the material is not described in Ph. Eur. (2) or a member state's pharmacopoeia.
The Guidance for Industry Container Closure System for Packaging Human Drugs and Biologics (1999) (3) covers all the packaging aspects for drug products intended for human use. The risk analysis is based on the different pharmaceutical forms and route of administration. All aspects of the packaging, not only the plastic, are considered. The requirements are based on the protection, safety, compatibility, and performance of the packaging. The toxicological aspect is considered with the USP biological reactivity testing (〈87〉 and 〈88〉) (8). The extraction is done with solvents recommended by the USP or selected following the user's risk analysis. Sometimes the extraction will be done after contact with the final product itself or a placebo. This approach is similar to the approach used in the food industry. The extractable study for orally administered products is performed retrospectively in case any impact on the quality of the plastic or the physicochemical characteristics of the final product occurs. Food regulation should be considered for products administered orally.
In Europe, specifications for leaching substances are based on a positive list of additives, under defined conditions of use. The general rules states that materials cannot release more than 10 mg/dm2 or 60 mg/kg if the container's nominal capacity lies between 500 mL and 10 L and the surface in contact with the product cannot be estimated. Some simulating media can be used in order to assess the migration, (e.g., purified water, acid aqueous solution, ethanol in aqueous solution, olive oil solution).
There is no general specification in the US as there is in Europe, but the approach is quite the same. Limits are based on an extraction with simulating solvents (distilled water, ethanol in aqueous solution).
Pharmacopoeias describe monographs or testing for some polymer materials or packaging. For plastic materials, Ph. Eur. provides a list of authorized additives and their limits (2). USP also describes biological reactivity testing, in vivo and in vitro testing (8), and the JP (9) and ISO 109993 (Biological evaluation of medical devices) provide testing defined in accordance with the contact type and duration (10).
Migration Phenomena Evalutation
When compatibility between a material and its content must be evaluated, it is necessary to have a good knowledge of the material in contact with the product and the physicochemical characteristics of the product. In fact, several mechanisms for the container–content interaction may occur between the material and the product, as seen in Figure 1, which shows
-
the migration of the product (content) into the container, with the
-
1a. adsorption (a surface action)
-
1b. absorption (penetration of active molecules into the material)
-
1c. permeation (actives molecules crossing the wall of the container)
-
-
the migration of the container components into the content, with the release of the container components (2)
-
the permeability: gas or liquid transfer phenomena through the wall of the container (3). Note: This aspect must be considered for semi-permeable materials (e.g., plastic bags).
The following consequences of these interactions may occur:
-
decrease of the activity due to an adsorption of the active substance on the material
-
active ingredient degradation due to released substances
-
content precipitation
-
pH change due to a leaching of the material components
-
appearance change (color) due to a leaching of the material components
-
analytical interference during the determination of the active ingredient
-
safety change due to a leaching of the material components
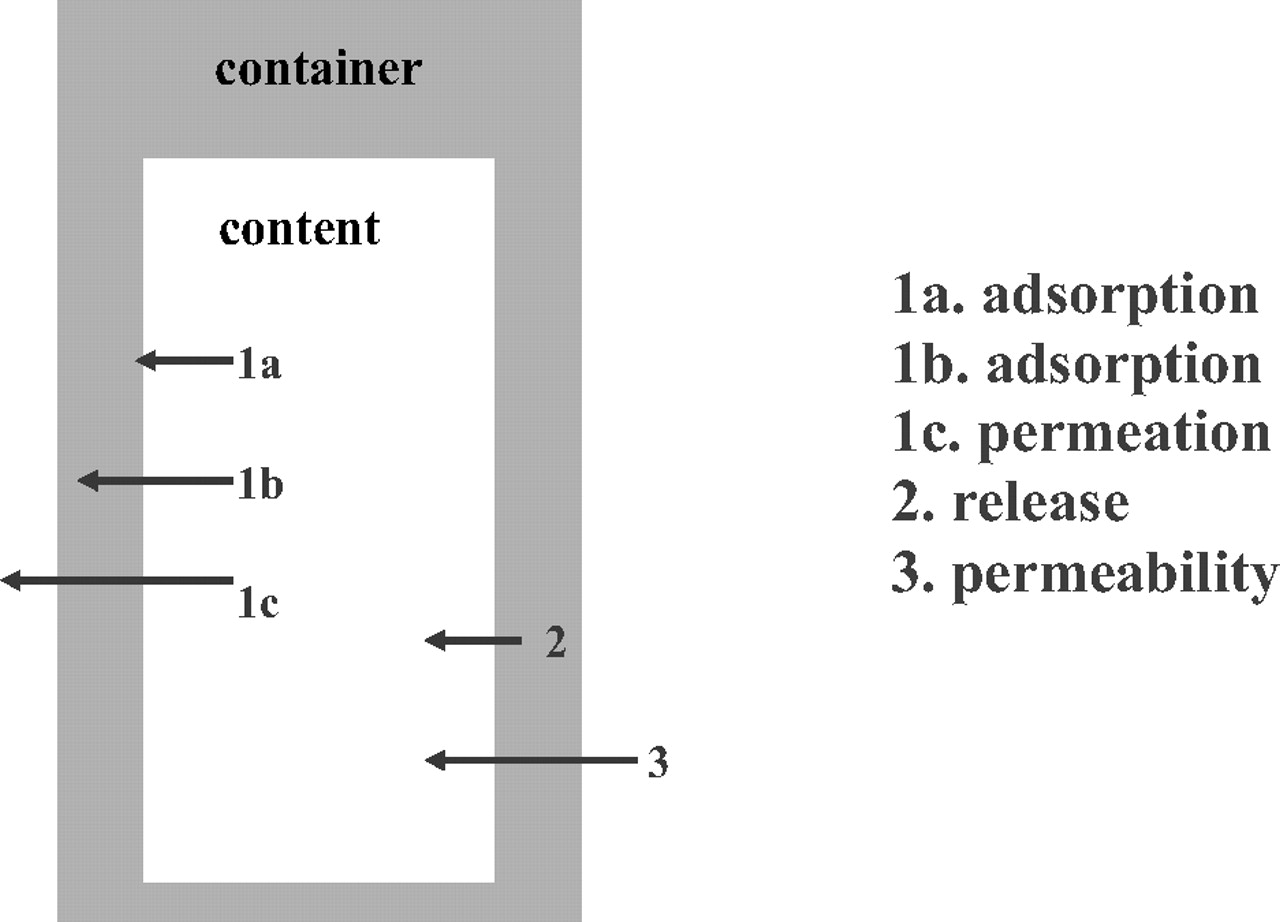
Migration phenomena.
All pharmaceutical products may be prone to interactions with a material, but the level of this interaction depends on the form (liquid, solid, etc.) and the route of administration (oral route, parenteral route, etc.).
Contact and interaction with a product, including printing inks and label adhesives, may occur at any time during storage (container) and during the manufacturing process (e.g., tubing, filters). Among all of the different materials used in the pharmaceutical industry, plastics and elastomers are the most critical because of their composition and addition of several components.
Glass and stainless steel containers or materials meeting EP or USP requirements may also leach some components, but according to the standard classification (e.g., USP type I borosilicate glass or stainless steel 316L) their quality guarantees the safety of these components.
The potential release of components may induce a decrease of pharmacological activity or a pH change. This interaction may be evaluated during the stability or validation study. The release of components depends on the solubility of the extractable into the polymer and its diffusion through the polymer. The solubility depends on the morphology of the polymer, and the extractable's temperature, structure, and molecular weight (MW) as well. Crystalline sites form a barrier for the migration, and migration is lower in semi-crystalline polymers. If the temperature increases, the structure of the crystalline sites then changes, and the crystalline barrier decreases as a result. In general, extractables with a low MW are very soluble, for example, butylated hydroxytoluene (BHT) and stearic acid. Extractables with a high MW are not very soluble (phenolic antioxidants, phosphate antioxidants). Furthermore, the solubility of the extractable depends on the level of exposure to media, and even high-MW extractables can be very soluble when they come in contact with organic solvents.
Diffusion is related to the temperature, because of Fick's law:
 where D is the diffusion coefficient related to the temperature
where D is the diffusion coefficient related to the temperature
 Diffusion is inversely proportional to the MW. Oligomers have a very limited diffusion (e.g., BHT MW 220: high diffusion; phenolic antioxidants MW 1010: low diffusion). Diffusion depends on the melting point or the glass transition temperature (Tg) of a material. If this Tg is low, the diffusion is high. Furthermore, the diffusion depends on the nature of the formulation (hydrophobic or hydrophilic liquid).
Diffusion is inversely proportional to the MW. Oligomers have a very limited diffusion (e.g., BHT MW 220: high diffusion; phenolic antioxidants MW 1010: low diffusion). Diffusion depends on the melting point or the glass transition temperature (Tg) of a material. If this Tg is low, the diffusion is high. Furthermore, the diffusion depends on the nature of the formulation (hydrophobic or hydrophilic liquid).
Toxicological Aspects
The evaluation of an interaction between a material and a product should also be based on toxicological criteria; such criteria are useful for developing threshold limits. An identification and quantification of potential extractables could be done, with the definition of a quantification threshold (QT), based on the intended use of the material and the product (contact duration, route of administration, dosage, and treatment duration) (11, 12). Threshold limits must preferably be based on scientific data published in the literature or in databases admitted by the scientific community.
The systemic toxicity, mutagenicity, carcinogenicity, teratogenicity, and intradermal reactivity for extractables described in the literature must be checked in order to avoid the need to use animal testing.
Several databases are available from the National Library of Medicine (NLM) and the National Institutes of Health (NIH) in the US and from the National Institute of Research and Safety (INRS) in France. The Hazardous Substances Data Bank (HSBD) and Integrated Risk Information System (IRIS) are open-access databases available on the NLM's website. IRIS is based on toxicological information collected by the Environmental Protection Agency (EPA).
Defining the no observed adverse effect level (NOAEL) or the lowest observed adverse effect level (LOAEL) is a fundamental step in a risk assessment methodology. The LD50 (lethal dose 50%) can also be used, but carefully, because of the sensitive transposition to humans. A toxicological evaluation should be conducted on identified extractables present at a level above the QT, with a definition of a safety concern threshold (SCT).
For extractables with no assessed health risks, several reference documents including the JP (9), USP (8), American Society for Testing and Materials (ASTM) (13, 14), and ISO Standards (10), propose methodologies covering all the risks associated with container use.
In vitro tests should always be considered before moving to animal testing. Cytotoxicity tests may represent a rational, extremely sensitive approach for the determination of the container compatibility with a pharmaceutical content. The test may be conducted in accordance with ISO 10993-5 or USP 〈87〉 for a qualitative approach, or with the Japanese Pharmacopoeia, for a qualitative and quantitative approach.
Decision Trees
We have developed an approach based on the safety of the product making contact with a material during storage or manufacturing process. The impact on the efficacy of the product should be evaluated through stability or validation data, with relevant tests on degradation products, material aspect/functionality, properties and performance, and active ingredient determination.
In order to assess the product's safety completely, it is necessary to be knowledgeable of the material and its manufacturing process. This is necessary because the migration depends on the material component's levels of solubility and diffusion through the material, as well as its MW. The material supplier holds this information and is able to provide the potential extractable and toxicological data.
The evaluation of the supplier data is the second step. According to the regulatory requirements, the supplier must provide the user with these data. Obtaining the exact qualitative and quantitative composition of the material from suppliers may be not very easy. In the US confidentiality issues can be addressed through the Drug Master File (DMF) or a Confidential Disclosure Agreement (CDA). The CDA is signed by both parties directly involved. It defines the aim of transmitting this confidential information, and it specifies who can have access to the information as well as the duration of access.
If materials are made of chemically and physically well-characterized substances, recognized in the published literature, and are based on their intended use, then a scientific judgment is necessary to determine which tests are required for the safety demonstration and consequently submitted to the authorities. The ISO 10993-1 implementation will generate adequate biological data to meet regulatory requirements.
Knowing the material characteristics (including the extractables, and the product properties) makes it possible to evaluate the amount of leachables and perform an evaluation of the individual toxicological risk. But in this case, the potential synergy between the different components cannot be evaluated. However, as part of a global toxicological evaluation of the final material, biological reactivity testing should be performed to provide more precise information on the impact of the material intended for use (e.g., after a treatment, such as sterilization). Cytotoxicity testing requested by the JP, USP, or ISO 10993 to determine the biological reactivity of materials can be conducted on the product after contact with the material.
Figure 2 presents an overview of this approach which uses the material (described in a pharmacopoeia or not) and the route of administration as entry points.
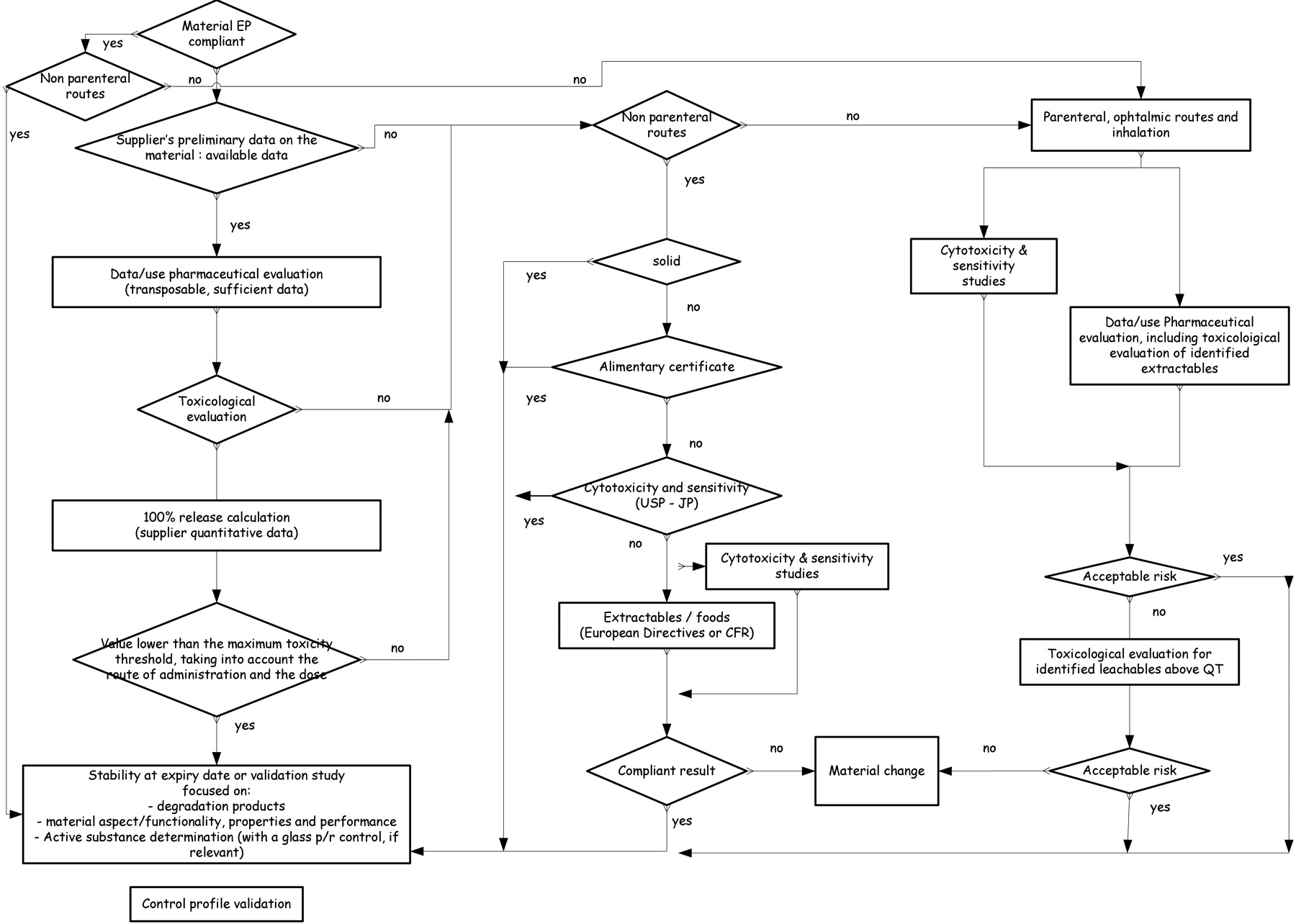
Decision tree.
If a non-parenteral-route material complies with a pharmacopoeia, then a stability or validation study must be initiated to detect changes in the material's appearance, active ingredient adsorption, or constitution of new impurities due to leaching. If the material is not described in a pharmacopoeia, some preliminary data related to the extraction profile and safety becomes necessary:
-
material composition and source: type of plastic (references), additives (qualitative and quantitative), process
-
biocompatibility (in vitro, in vivo tests), according to USP 〈87〉 and 〈88〉 or JP or ISO 10993
-
physicochemical tests, according to USP 〈661〉, 〈381〉, Ph. Eur. or food monographs
-
technical characteristics of the material (flexibility, permeation, performance)
-
certificate of analysis, internal specifications, analytical methods
-
potential extractable list and analytical methods
Based on these data, a pharmaceutical evaluation should be done (taking into account the intended use of the material). A toxicological evaluation should also be included. The 100% release level can be calculated and compared to the maximum toxicity threshold related to the product.
When these data are not available, incomplete, or not easy to use, a more detailed analysis that depends on the route of administration must be performed. For a non-parenteral-route, liquid, or semi-solid forms, the availability of an alimentary certificate should be verified, as well as the availability of cytotoxicity and sensitivity tests. Without these results, the manufacturer can choose to launch either cytotoxicity/sensitivity tests or extractables studies, according to food standards, along with a qualitative and quantitative analysis. Where the cytotoxicity tests or food compatibility tests are non-compliant, it would be better to change the material. In all the other cases, as well as for solid forms, a stability or validation study is sufficient to highlight an interaction.
For a parenteral route, compliance with Ph. Eur. is not sufficient to avoid further studies. A pharmaceutical evaluation should be done in addition to cytotoxicity tests by placing the product in real conditions (based on a risk analysis). The pharmaceutical evaluation should include
-
an evaluation of the migration phenomena
-
an evaluation of the intended use of the material
-
an identification of extractables above a QT if there are no structural alerts for genotoxic or carcinotoxic component. If there are structural alerts, all the extractables must be identified.
Identification of leachables and a toxicological evaluation for identified leachables above the QT must be conducted according to the pharmaceutical evaluation's risk analysis and the cytotoxicity results.
Extractables Study
Extraction studies will allow for identification and quantification of extractables above an identification threshold, based on the information provided by the supplier. Another objective of the extraction studies is to develop analytical methods to quantify leachables, if needed.
Material is exposed to an appropriate solvent system at extreme conditions in order to maximize the amount of extractables from the material in the solvent. The use of multiple extraction solvents should be evaluated according to a documented rationale based on actual process fluid composition, to provide adequate assurance that no extractable is missed. The most common extraction technique is refluxing (Soxhlet extraction). Some alternative techniques may be used, such as a less aggressive temperature, accelerated solvent extractor, sonication, or extraction in a sealed container placed in an oven or autoclave.
If the objective is to validate the material for a specific product, then either a placebo or the final product may be used. If the objective is to validate the material for several products, then several media, representative of all the products, may be used, for example, sodium chloride solution (NaCl) 0.9%, purified water, ethanol, polyethylene glycol 400 (PEG 400). The resulting solution is then analyzed with advanced instruments to identify and quantify extractable substances. The instrument methods used must be able to identify volatile substances, metallic and non-metallic components, and elements.
The following multiple analytical techniques are used:
-
High-performance liquid chromatography: photo-diode-array-detection combined with mass spectrometry (HPLC/DAD-MS)
-
Gas chromatography–mass spectrometry (GC/MS) for organic analysis
-
Head space gas chromatography: mass spectrometry (HS-GC-MS) for volatile components
-
Capillary zone electrophoresis (CZE) or micellar electrokinetic chromatography (MEKC) for solvent-sensitive components or complexes
-
Inductively coupled plasma mass spectrometry (ICP-MS) for metal analysis
-
Newer analytical methods including LC/NMR (liquid chromatography/nuclear magnetic resonance spectroscopy) and SIMS (secondary ion mass spectrometry) can be used.
These methods must be validated at least for the limit of detection (LOD) and the limit of quantitation (LOQ) in order to determine the quantity of extractables released (15). The ratio between the surface area of the material and the volume of the content in the extraction conditions is calculated in order to correlate with the real-use conditions.
If extractables are detected and identified, then their toxicity must be investigated based on the toxicological databases, and a maximum limit allowed for toxicological safety must be defined according to the intended use (ICH Q3B guidelines) (16) in order to determine which compounds need to be tracked as potential leachables in the product. The need to track representative extractable compounds in routine quality control screening (quality control methods for supplier materials) should be evaluated.
Leachables Study
This study is conducted under normal storage conditions with the product. The objective is to detect and quantify the components that have been identified during the extractables study and that can be released over time. Methods must be validated for the matrix of the product in order to provide sufficient sensitivity, accuracy, precision, and linearity within a range, and to allow the possibility to quantify the leachables.
These studies should be conducted at least at the beginning and the end of the shelf life, on three batches of material, and as a control without contact with the tested material, using real-time conditions and accelerated conditions (if applicable). An additional intermediate time-point can be added, for example, at mid-shelf life. If leachables are detected and identified, their toxicity must be investigated according to the maximum limits admitted for the intended use of the material and the product.
Conclusion
In conclusion, the container–content interaction study is essential to guarantee the quality of a pharmaceutical product in contact with a material. However, this study can be conducted in different ways, depending on the route of administration and the contact duration.
In any case, the following points should be considered:
-
The supplier data should be available and must be evaluated by the user according to the use of the material, its constitution, and the physicochemical properties of the product.
-
Biological reactivity tests, such as cytotoxicity tests, along with a definition of allowable limits, should be done in real conditions to determine the global non-toxicity without individual separation.
-
A toxicological evaluation for each identified extractable above its QT should be done in order to guarantee the safety of the product making contact with this material
-
The leachable studies should be performed when extractables above their QT have been identified, or if the identification of one component of the material responsible for an interaction is requested in order to select a new material.
-
A stability study for packaging materials or a validation study for material used during the manufacturing process equipment must be conducted in order to evaluate the impact of the material on the quality and efficacy of the product (17, 18).
-
According to the evaluation of the supplier data and the biological reactivity testing under actual conditions, the relevance of the toxicological evaluation of the individual extractables and leachables must be considered.
Footnotes
- © PDA, Inc. 2009



{kind=link}
{kind=link}