
Abstract
The manufacture and supply of pharmaceutical products can be a very complex operation. Companies may purchase a wide variety of materials, from active pharmaceutical ingredients to packaging materials, from “in company” suppliers or from third parties. They may also purchase or contract a number of services such as analysis, data management, audit, among others. It is very important that these materials and services are of the requisite quality in order that patient safety and company reputation are adequately protected. Such quality requirements are ongoing throughout the product life cycle.
In recent years, assurance of quality has been derived via audit of the supplier or service provider and by using periodic audits, for example, annually or at least once every 5 years. In the past, companies may have used an audit only for what they considered to be “key” materials or services and used testing on receipt, for example, as their quality assurance measure for “less important” supplies. Such approaches changed as a result of pressure from both internal sources and regulators to the time-driven audit for all suppliers and service providers. Companies recognised that eventually they would be responsible for the quality of the supplied product or service and audit, although providing only a “snapshot in time” seemed a convenient way of demonstrating that they were meeting their obligations. Problems, however, still occur with the supplied product or service and will usually be more frequent from certain suppliers. Additionally, some third-party suppliers will no longer accept routine audits from individual companies, as the overall audit load can exceed one external audit per working day. Consequently a different model is needed for assessing supplier quality.
This paper presents a risk-based approach to creating an audit plan and for scheduling the frequency and depth of such audits. The approach is based on the principles and process of the Quality Risk Management guideline (ICH Q9) of the International Conference on Harmonisation (ICH). It proposes that if regulatory conditions allow, it may be possible to remove the need to conduct audits on the sole basis of time elapsed since the last audit, or at least to increase the time interval between such audits without compromising either patient safety or company reputation. The proposal is equally applicable to both large and small companies. Small companies may find it particularly useful in cases where they use a supplier that may have a monopoly position or that serves many other pharmaceutical companies. In such circumstances the supplier may be reluctant or even refuse to accept audits from some individual companies because of their low purchasing levels. A similar approach could be proposed for regulatory authorities for the scheduling of regulatory inspections.
1. Introduction and Scope
The Quality Risk Management guideline ICH Q9 provides the opportunity to implement the risk management principles to the audit and inspection processes as part of an integrated Quality Management System (ICH Q9, Annex II.1).
By definition an “audit” is conducted by a company and an “inspection” by health authorities or a recognised certification body; however, there is little difference in the overall processes performed. The set-up of an audit or inspection plan is a key initial process step, particularly where there is potentially a large number of audits or inspections to be performed. This paper outlines a risk-based approach to preparing such a plan. It does so in a manner that limits the number of audits or inspections and their duration, depth, and frequency. This is achieved without consequently raising the risk to patients arising from good manufacturing process (GMP) issues, using the Quality Risk Management methodology and principles as described in ICH Q9 plus the regulatory framework (e.g., in the European Union [EU]: Eudralex Vol. 4 [EU-GMP] Part 1, Chapter 1, and Part 2).
This paper gives a proposal for the practical implementation of Quality Risk Management principles in developing and documenting an annual audit plan. These principles are used to summarise the knowledge gained from audits conducted and from additional means of assessing compliance status (see Section 3.2). Multi-factorial risk assessment is used to develop the plan, to prioritize sites for auditing, and as a means of assessing the required depth of focus and duration of the audit. This process can be applied in principle to all kinds of “suppliers” either from the company's own manufacturing sites, its contractors or licensees, and third-party materials or service providers, for example
-
Active pharmaceutical ingredient (API) starting material
-
APIs (small molecule/biotech)
-
Excipients
-
Packaging materials
-
Bulk
-
Drug (medicinal) products
-
Contract manufacturers and laboratories/service providers
The process can be used at local sites as well as for global auditing operations. It can also be applied, by adapting the categories, to drug safety or pharmacovigilance audits.
Regulatory inspectorates could use a similar approach for scheduling or conducting regulatory inspections in their country, collective of states or countries to which a common regulatory code applies, for example, the EU, World Health Organisation (WHO), Cooperation Council for the Arab States of the Gulf (GCC), Association of Southeast Asian Nations (ASEAN).
The two most common drivers for audit and inspection scheduling are time (e.g, not more than 2 years since the last audit or inspection) and/or new product introduction. Such requirements may arise from country-specific rules or legislation. The implementation of Quality Risk Management principles may facilitate a change in such drivers in the future.
2. The Auditing Process
Companies and/or regulators need to gain knowledge of the compliance status of the supplier with respect to GMP/GDP (good manufacturing practice/good distribution practices) and/or contractual/regulatory requirements in terms of the expected quality of supplied goods or services. This knowledge is usually determined by an audit or inspection and is a significant factor in developing confidence in the quality of the supplied product or service.
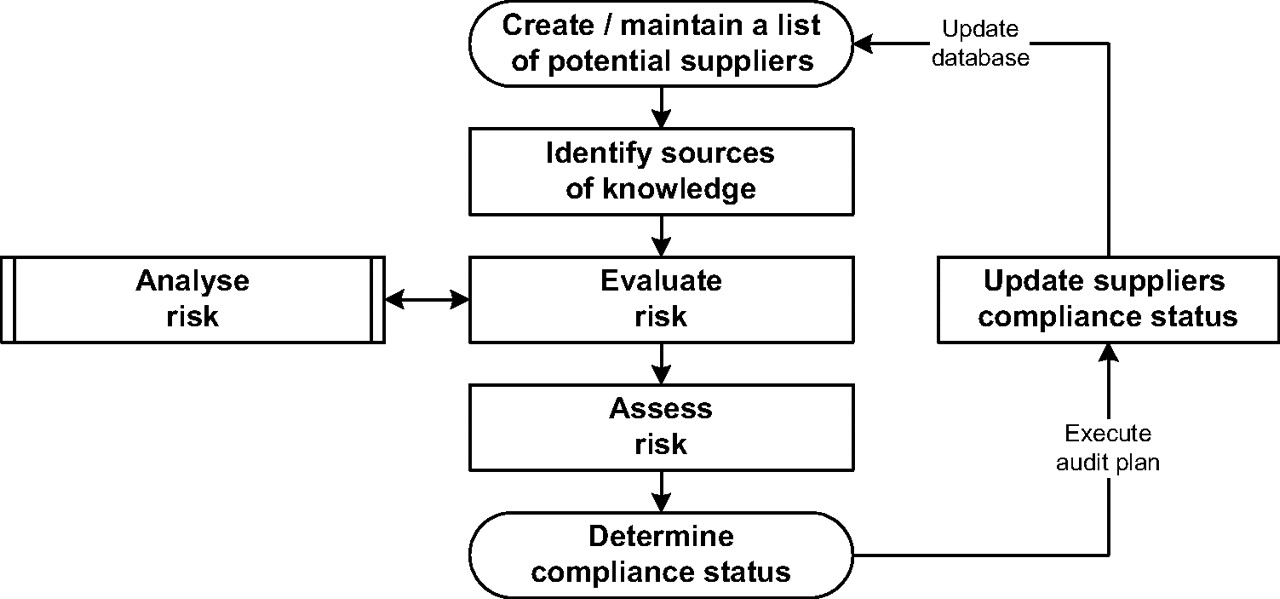
The principal process steps for “audits” conducted by industry or “inspections” conducted by regulators can be described as a sequence of gathering data, analysing information, and evaluating knowledge for decision making (Figure 1).

General process flow of an auditing/inspection process.
Once a potential supplier is identified, a decision whether or not to schedule an audit has to be made. In order to make this decision, accessible sources of knowledge about the supplier should be identified and evaluated. For example:
-
A new product is being introduced onto a site or manufacturing operation and similar products are already handled at that site; the GMP status for those products is known to be satisfactory.
-
The supplier may have been recently assessed by a trusted assessor for similar operations, and information regarding that assessment is available.
In such cases it may be possible to deduce the compliance status of the supplier for the new proposal and to commence supply without scheduling an audit. It is important to ensure that the information used to consider the supplier status is relevant to the new product or service being provided.
If there is no current satisfactory data available, then an audit will be scheduled, planned, and conducted, and any observations will be summarised in the audit report. The timely response of the supplier to observations and/or recommendations within the audit report is assessed and used to determine the status of the supplier and to make the final accept/reject decision. The evaluated status may be based on the audit observations, the response and action taken, the result of any re-audit, and any other factors deemed relevant.
Ideally, local and company GMP audit or quality assurance (QA) groups will use some form of an audit evaluation scheme for sharing GMP compliance status of an internal (own site) or an external supplier (contractor, licensee) with senior management. The compliance status report generates an overview of the knowledge of this supplier (Table I) and usually has some form of ranking, e.g., “good”, “satisfactory”, or “unsatisfactory”. One simple example of a ranking method is a traffic light system: green = “good”, amber = “satisfactory”, red = “unsatisfactory”.
An Evaluation Scheme of Audit and Inspection Results for Sharing Knowledge (e.g., to Senior Management in Companies)
Following discussions with colleagues within the pharmaceutical industry, the authors are aware that many companies internally rank inspections conducted on their suppliers by regulatory authorities using the same system and then communicate the output to senior management. The communicated information is in turn used in enlightened companies to help it fulfil its Quality Management System obligations in terms of management responsibility and continual improvement. It should also form a key element in the operation and decision making elements of any corporate sourcing strategy. Senior management use the knowledge gained from this process to assist in decision making as to whether or not to continue working with a particular supplier. Where the supplier involved is a part of their own company, senior management can have a direct impact in assuring that appropriate and timely corrective measures are taken in order to help protect patient safety and company reputation. This is seen as part of “Management Responsibility” within the company (ICH Q10) or an International Organization for Standardization (ISO) Quality Management System.
In the EU the QP (qualified person) will have a particular interest in the process, as it is one of the key means by which QPs assure themselves of the GMP status of suppliers, goods, and services. The knowledge gained from the audit evaluation process can also be used in the audit plan to schedule the timing, focus, and depth of the next audit. Where information from an audit or inspection is inadequate with respect to a given supplier, it would be normal to schedule a follow-up audit immediately. Quality Risk Management provides opportunities to rethink and refocus resources in order to protect patients and not waste time and resources conducting poorly focused or unnecessary audits.
3. The Process of Establishing a Risk-Based Audit Plan
The risk assessment and prioritisation of individual suppliers for auditing is primarily the responsibility of a quality unit, and the process will usually be defined in a standard operating procedure (SOP). The head of the quality unit or his/her nominee ensures compliance with regulations, approves the annual audit plan, and also approves any additional audits outside of the scope of the plan, based on business or compliance needs. Allowance must be made for additional audits, for example, audits triggered by factors such as new quality information, production or product problems or recalls, commercial needs, and so forth.
The audit plan must be in line with current regulatory requirements for auditing frequencies for supplied markets. Such regulatory requirements will themselves be subject to continual change as the authorities build Quality Risk Management principles into their procedures.
3.1. Create and Maintain a List of (Potential) Suppliers
An individual working in the Quality Unit is responsible for maintaining a list or database of potential suppliers. Additions or deletions to the list may arise as a result of initiatives from other parts of the company, e.g., research and development, supply chain management, contract and licensee management. It is vital that such proposed changes are communicated in a timely manner to the individual in QA with responsibility for maintaining the list. A short, precise rationale for such changes should be recorded as appropriate. The level of recording of such change should be in accordance with the principles of ICH Q9.
3.2. Identify Sources of Knowledge about Suppliers
Relevant sources of knowledge about a supplier are identified. The prime focus will concern GMP status, although other information such as business stability, reliability of supply, capacity, or existing supply to the pharmaceutical industry are very relevant. Valuable information on non-GMP issues can often be found in the company's annual report. GMP information may be gained, for instance, via access to audit reports from third parties (e.g., other companies, consultancies, assessment bodies) that are trusted by your company or, where appropriate, from regulatory inspection information.
Regulatory authorities may share conclusions and other information from inspections via legal agreements such as Mutual Recognition Agreements, Memoranda of Understanding, and Confidentiality Agreements. They may also use other information-sharing mechanisms via organisations such as PIC/S (Pharmaceutical Inspection Co-Operation Scheme). Such measures may result in the avoidance of duplicate inspections by different inspectorates. Industry may also gain access to GMP status reports from inspectorates via vehicles such as the European Medicines Agency European Union Drug Regulatory Authorities (EUDRA) GMP database.
Care should be taken not to overcomplicate this process through a desire for standardisation. The requirements for each supplier should be appropriate to the risk presented by that supplier (see Section 3.3). Again the level of effort (in this case audit) should be in accordance with the principles of ICH Q9.
The process used to determine and maintain the compliance status is shown in Figure 2.

Process flow to determine and maintain compliance status.
3.3. Analyse and Determine Risk of an Individual Supplier
The determination and analysis of risk factors is only performed once for each supply scenario to set up the approach. However, it may be necessary to make changes to the approach in light of new challenges or experiences or change of scope regarding the services provided.
The ICH Q9 guideline (ICH Q9, Annex II.1) gives a list of 12 factors to facilitate the process of defining the frequency and scope of audits. Since publication of the ICH Q9 guideline, other factors have been highlighted by commentators that are considered to be important in a particular supply situation. It is impossible for industry and regulators to have an exhaustive list of individual factors with all data for every supplier. The factors used to assess a supplier must ultimately be considered on a case-by-case basis using risk-based methodologies. Additionally, any mathematical calculation of such risk factors is vulnerable in that other unknown risks may exist. Therefore when such calculations are made, it is normal (and common sense dictates) to use assumptions and/or weighting of the factors, using previous experience gained from using these factors or similar suppliers.
Quality Risk Management provides the opportunity to group these factors by answering the three basic questions given in ICH Q9 (Chapter 4.3):
-
What might go wrong?
-
What is the likelihood (probability) it will go wrong?
-
What are the consequences (severity)?
The ability to detect a potential GMP and/or regulatory noncompliance (detectability) is also a factor in the estimation of the underlying risk. By assessing these guiding questions the factors can be grouped into four categories:
-
Severity: Compliance
-
Severity: Availability
-
Probability: Complexity
-
Detectability: History
Using these simple categories allows for a high-level risk assessment but facilitates sufficient ranking of a potential supplier without having to use significant organisational resources or additional interfaces to determine more details.
Such an approach is commensurate with the second principle of ICH Q9: “The level of effort, formality and documentation of the Quality Risk Management process should be commensurate with the level of risk”. Ongoing decisions for any current supplier can be facilitated by the use of adequate knowledge management procedures and processes that will identify triggers for change in the risk assessment process. This could be a part of the system for corrective and preventive actions (CAPA) in the company's pharmaceutical Quality Management System. This risk assessment process systematically organises the information to support a risk decision. In each specific context the assessment is performed against a predetermined set of risk criteria.
The “potential for risk” (risk level) is assessed semi-quantitatively for each category. A four-level numerical approach (1–4) is recommended. Other ranking levels can be used (e.g., 1–5, 1–10), but the four-level approach avoids a middle value such that the risk levels are defined as
-
Extremely High
-
High
-
Low
-
Very low
This approach of weighted risk factors is further explained in Section 3.4.
Category: Compliance:
This category describes the GMP and regulatory compliance status of a supplier. This represents the severity factor according to the Quality Risk Management definitions given in ICH Q9. It relates to the protection of the patient by assuring that safety and efficacy of the product are not compromised by inadequate GMP or noncompliance with chemistry and manufacturing aspects of the regulatory filing. The severity rating may vary according to the type of product being manufactured and the criticality of that product within the company's product portfolio.
The risk criteria “compliance” classifies the GMP and regulatory compliance status of the supplier using all available information from company knowledge management systems to provide key information, including
-
GMP compliance metrics
-
History of issues and recalls
-
Results from previous audits and inspections (including the suppliers responses to audit or inspection observations)
-
Results and trends from the quality testing of received goods
-
Changes to the suppliers management (e.g., takeovers), portfolio, facility, or ways of working since previous assessments that could affect its ability to supply
-
Projected changes in demand for the drug product, component, or service that the supplier may not be able to meet
Data for this assessment can be taken from different sources, for example, the previous audits, quality history, and information issued by the supplier such as annual reports. If the compliance status is unknown, the highest risk level is used (Table II).
Potential Impact Levels of the Category “Compliance”
Category: Availability:
The use of the “availability” risk criteria is linked to the ability of the supplying company to manufacture and release compliant starting material (API, excipient), intermediate, or drug product for sale or to provide a specific service such as analytical testing when required. It is also therefore ultimately linked to the availability of the drug product to the patient in the marketplace. Such a risk may arise from GMP, regulatory compliance, or capacity issues with the supplier. In general, for example, most drug product, API, or excipient manufacturing sites of a supplier would be given a high-risk ranking. Suppliers of raw materials for APIs that may be easily and legally substituted with another supplier would be given a low-risk level (Table III).
Potential Impact Levels of the Category “Availability”
The factors on the category “availability” are weighted low, as they describe factors that may influence compliance status but do not normally dominate the decision-making process. For regulators “availability” could be related to the importance of the drug on the local market, for example, a medicine for tropical diseases in a tropical market and/or the availability of legal alternatives within a specific market. Clearly, in such cases, the impact of potential supply risks needs to be fully assessed by the company, and appropriate mechanisms to maintain supply (inventory holding, contingency supply) need to be considered.
Category: Complexity:
The category “complexity” classifies the potential impact on the patient arising from complex patient operations, for example, where the patient may need to use a complicated delivery device such as an inhaler in order to take the product, or the potential impact on the product itself arising from the complexity of the process and/or the regulatory requirements.
The terms “complex” and “complicated” have different meanings. Complicated items are not necessarily complex. Some examples of complexity that may need to be evaluated include
-
Cultural aspects, e.g., of senior management, people, knowledge, education, risk management culture, staff turnover
-
Physical size of the supplier, for example, number of people, number of departments
-
Number of interfaces at a supplier, for example,
-
soft interfaces (e.g., diversity of organisational roles, management oversight, data or material transfer, separated responsibility, management of key performance indicators), degree of standardisation (e.g., of processes)
-
hard interfaces (e.g., development activities, API synthesis including special raw materials supply, milling, micronizing, formulation to bulk, drug product manufacturing, major packaging activities, major distribution activities)
-
-
Architecture of risks,” for example, managing risks, risk awareness culture, total disclosure culture (Note: a “total disclosure culture” requires that a company should communicate to interested parties all identified risks and ways in which it plans to manage those risks along with the timescales involved)
-
Stress factors,” for example, continuity and stability of organisation, product portfolio (continuity of product portfolio, new product introductions, supply chain processes, investigational medicinal product (IMP) manufacturing), reorganisations (downsizing, contracting out, moving to emerging markets), mixed scenarios (e.g., dedicated or multipurpose, market supply or IMP, aseptic or “normal” products, chemical or biotech, in-house or third-party business)
Another aspect of complexity is that arising from regulatory requirements. Examples might be
-
Multiple operational disciplines at the same site (e.g., human drugs, veterinary drugs, dietary supplements, food products, animal feed products).
-
Manufacturing operations such as those for biological products, sterile products, radiopharmaceuticals, or vaccines that are subject to complex regulations.
Complexity may be introduced as a consequence of the use of the material being manufactured (e.g., IMP manufacturing, drug products with local, regional, or global filing) or the service provided. Additional complexity may be introduced via the GMP standard required for the manufacturing process (e.g., dedicated or multipurpose facility, API [ICH Q7] for small molecules and biologics), drug product, bulk, final product, primary packaging, secondary packaging, storage operations, GDP activities).
It is a very difficult task to obtain, maintain, and evaluate these data from every supplier. Therefore it is unlikely that a model based on these factors would be successful without making a number of assumptions based on background knowledge using the second principle on Quality Risk Management as described in ICH Q9: “The level of effort, formality and documentation of the quality risk management process should be commensurate with the level of risk,” which avoids the use of over-complex approaches.
The focus should be on what complexity factors have the greatest potential impact on the quality of the product and the viability of the business (Table IV).
Potential Impact Levels of the Category “Complexity”
Category: History:
Many companies use the time since the last audit as the major, or even the only, trigger for scheduling an audit; some regulators adopt a similar approach. The risk criteria “history” applies a different emphasis based on up-to-date knowledge of the compliance status of the supplier. The information that may be used, if available, includes
-
Timing of and information gained from last audit, site visits, audits from third parties
-
Inspections or regulatory action by a health authority since the last audit
-
Changes made to the site ownership, infrastructure, product range or focus
This approach focuses on information that allows for the detection of quality risks. If regulatory positions allow, it may be possible to remove the need to conduct audits on the basis of the time elapsed since last audit, or at least to increase the time interval for such audits (Table V).
Potential Impact Levels of the Category “History”
3.4. Evaluating the Risk Arising from a Supplier
The risk factors identified for the four categories (compliance, availability, complexity, history) are evaluated considering the risk to patients and to the company. A weighted risk factor reflecting the categories is then assigned for each risk level. The weighted risk factors are related to the overall importance of each risk category (Table VI).
Weighting by Protection of Patient/Company
The risk factors for any specific risk level within the four categories may not always be the same. The authors have utilised the highest factors in the category of compliance because this is the main focus for conducting audits and in all probability represents the highest risk to patients and to company reputation. Note: There will always be debate on the weighting of specific factors. A team can spend hours deciding whether 10, 6, 3, 1 is “correct” or 10, 7, 3, 1 is more appropriate. In practice the results of the risk assessment provide an aid to decision making, rather than a noncontestable mathematical solution.
It is very important to always work in accordance with the following two rules:
-
In case of different activities with different rankings, always take the highest ranking (e.g., see Table IV on complexity: this supplier supplies the company with both sterile API [risk factor = 8] and nonsterile API [risk factor = 2]; risk factor 8 is therefore used in the calculation of the overall risk)
-
In cases where there is no information for a particular category, then the highest ranking should be used (e.g., 10 for compliance, 8 for complexity, 6 for availability and history)
Once the ranking for each category is complete for each individual supplier, that supplier's overall risk factor can be calculated by multiplying its four individual risk factors. Subsequently all evaluated suppliers can be sorted according to their overall risk factor. The value obtained is used to set auditing priorities by sorting companies according to their overall risk factor (Table VII). It must be remembered that in some countries there may be legislation that requires audits to be conducted within a specific timeframe, and companies must be in compliance with such legislation. Suppliers that under such legislation are not yet due for an audit should, however, continue to be put through the risk ranking process, as it may be demonstrated that they are in fact high risk and need to be scheduled for an audit before the proposed time-based audit interval.
Evaluating the Risk Arising from a Supplier Sorted by the Overall Risk Factor
3.5. Finalise the Audit Plan
In the initial stages of creating the audit plan it is recommended that all suppliers with total risk factors higher than 96 should be entered into the plan. This factor (96) has been chosen because it is the figure obtained by multiplying the factors for the acceptable level of each category:
-
Compliance = Factor 3
-
Availability = Factor 4
-
Complexity = Factor 2
-
History = Factor 4
The factors “3, 4, 2, 4” were selected by the authors as being representative of a level of risk that is just acceptable in terms of product quality using the right effort and formality (see ICH Q9 principles) in a general supply situation.
The calculated figure of 96 (3 × 4 × 2 × 4 = 96) should not be absolutely fixed; it is used as a guidance only. The audit program assessment panel will need to judge whether this is appropriate for the respective suppliers under evaluation and may wish to use other risk factors to provide an alternative warning limit with appropriate rationale.
There are no fixed rules in ICH Q9 concerning the definition of thresholds that result from the failure mode and effects analysis (FMEA) methodology (using risk priority numbers in risk evaluation). The ranking system has to be good enough to aid decision making and prioritisation, but the final risk decision is the responsibility of the company or regulator.
Audits may be scheduled for suppliers with a score of less than 96, and some suppliers with a higher score may not have an audit scheduled. This is consistent with ICH Q9 as long as the rationale for such decisions is justifiable and is recorded. It is recommended that the rationale be included within the audit plan as part of the risk management process.
It is possible to streamline the method of calculation in such a way that only the severity (i.e., compliance) and probability (i.e., availability, complexity) factors are used in an initial calculation. Then, for example, if a traffic light system is being used, any yellow or amber results are further assessed by making use of the history criterion (this might particularly relate to detectability) in order to see if the risk then becomes red (higher) or green (lower).
It is worth remembering, particularly within a large company, that audit plans are made by many other departments, for example, safety/health/environment, development, purchasing/business, and finance. It is logical in such circumstances to check synergies with the needs from other sites and departments and where possible to consider making joint audits. Such an approach can maximise the benefit for both your company and the supplier.
As has been previously indicated, the higher the complexity the greater the risk. Therefore, the more complex the auditor's business the greater will be the need for inclusion of the audit plan process within the Quality Management System. Fundamental aspects will be the need for an SOP describing the audit plan development system, clarification of roles and responsibilities, sign off, and publication of the audit plan. Links to the Change Management System will be needed for the addition or deletion of audits and suppliers within the plan.
3.6. Operational Tips
The potential supplier matrix (list of potential suppliers) can be set up as a spreadsheet or a database that lists all the sites, contractors, licensees, and suppliers that are within the scope of the respective organisation. Within the spreadsheet or database, a section for risk assessment can be incorporated. An overall risk factor, used to set auditing priorities, is automatically calculated for each potential supplier, taking into consideration compliance status, strategic interests, complexity of products and processes, and audit history.
The architecture of the spreadsheet or database can be summarised as
-
Risk initiation section: the listing of all potential suppliers including type of site, business partner name and location
-
Risk assessment section: the allocation of discrete values (1–4) for each risk criteria plus the rationale for assigning the individual weighted risk factors for each of the risk categories. In addition, these sections should also record the initials of the individual managers responsible for carrying out and approving the assessments. The overall risk factor is then determined by an automatic spreadsheet calculation.
-
Audit planning section: the date of the last audit (if known), the specified audit frequency according to legal requirements, and the calculated date (month and year) by which time the next audit should take place.
-
Comment section: used for rationales, explanations, and clarifications.
4. Experience with This Procedure
This way of scheduling audits has been implemented in F. Hoffmann-La Roche global quality as a large multinational company since 2006. Initially it was used for scheduling global audits of drug (medicinal) product suppliers and excipient suppliers. The number of audits performed have been reduced by approximately 40% from a purely time-based schedule without any detectable downside to product quality or patient safety. The resources that have been saved have been used for additional training and for facilitating the sharing of best practice approaches among the company's own sites and also with contractors. It is our experience that approximately 90% of external suppliers have been willing to provide both inspection and audit reports plus their own responses as relevant sources of knowledge about their company when this system has been used. These documents have, in turn, been assessed and conclusions drawn to rank the suppliers. In a very few cases, a short audit (1 day) was scheduled to check the performance of the implementation of the proposed measures. This procedure was at all times welcomed by the supplier.
There is no indication of an increase of quality problems, for example, no upward trend in deviations, investigation, complaints, issues, or recalls.
5. Conclusion
This paper describes a risk-based process for the development of an audit plan that is driven by factors other than time since the last audit. It also recommends that all audits within the plan need not be of the same duration or depth of focus. The process uses a robust framework to assess GMP status and wider supply capability of a particular “in house” or external supplier of products or services from relevant and trusted sources of knowledge about that supplier. Individual risk ratings, which always default to the highest value where knowledge is lacking, are then used in a simple mathematical calculation. The output from this calculation gives a measure of the risk that a particular supplier ultimately presents to the patient, product, and the supplied company's reputation. The higher the risk, the greater the need to focus resources of both the supplied company and the supplier toward reducing that risk to an acceptable level. The approach is based on the principles and process of ICH Q9 (Quality Risk Management).
Although the paper focuses on the audit plan for a company and its suppliers, a similar approach could be used for regulators and regulated companies. Both scenarios would, however, need to be in compliance with all legal requirements relevant to that scenario.
Small companies may find that certain suppliers are reluctant or even refuse to accept their audits because of their low usage or low value of their purchases. If the supplier has a monopoly position or serves many other pharmaceutical companies, then as long as legal requirements are met the “small” company may assess the supplier status without a direct audit. GMP compliance and other relevant information may be gained about the supplier and applied via the risk-based audit plan system (see also Section 3.2).
As previously mentioned, the described process is already being used by F. Hoffmann-La Roche global quality to determine the frequency of internal and external supplier audits wherever the legal requirements allows. The South African Health Authorities has indicated at conferences that they also use a similar approach with respect to determining the need for conducting foreign inspections.
Other approaches are currently being discussed and trialed for scheduling and/or conducting regulatory inspections by, for example, the Australian Therapeutic Goods Administration (TGA), UK Medicines and Helathcare products Regulatory Agency (MHRA), European Medicines Agency (EMEA) (for products registered by the centralised procedure only), US Food and Drug Administration (FDA), and Japanese inspectorates. These processes differ and could include additional time-consuming paperwork to be supplied from company to regulator. Harmonised approaches such as using the PIC/S Site Master File format (1) or the WHO Certification of Pharmaceutical Product (CPP) (2), for example, for recognising the ICH Q7 standard for APIs rather than a different format for each inspectorate would be welcomed by industry in the future.
Acknowledgments
We thank the topic team on foreign GMP inspections of the European Federation of Industry and Associations (EFPIA) for supporting these discussions as well as the discussions after presenting initial thoughts on this model at PDA conferences.
- © PDA, Inc. 2009




{kind=link}
{kind=link}