
Abstract
Raw materials are critical components of product manufacture; these include source materials such as cell substrates, tissues, and biological fluids required for product manufacture, as well as biological materials required for cell growth, propagation, differentiation, and selection. Adventitious viruses are a major safety concern in biological raw materials. This paper discusses the specific concerns related to different types of biological materials and presents the Center for Biologics Evaluation and Research's perspective on the qualification and management of raw materials for purposes of developing a safety program for the manufacture of biological products.
Introduction
Safety is paramount in biologics and involves rigorous qualification and management of the various biological raw materials that are used at different stages in product manufacture. This presentation pertains to various Center for Biologics Evaluation and Research (CBER) products regulated by the different offices: vaccines, toxoids, and allergenic extracts (for allergy shots and tests), in the Office of Vaccines Research and Review (OVRR); somatic cell products, gene therapy products, as well as human tissue and cellular products for transplantation, in the Office of Cell Therapy and Gene Therapy (OCTGT); and blood, blood components, anti-toxins, anti-venoms, immune globulins, immune Fabs, and medical devices and test kits, in the Office of Blood Research and Review (OBRR). These products can be grouped into two broad, general categories based upon the degree of virus inactivation and removal (i.e., clearance) during manufacture: (1) products that are not inactivated and are minimally purified and (2) inactivated products that may have virus clearance and some purification steps. The non-inactivated products include live, attenuated, and vector-based virus vaccines, allergenic extracts (such as pollens, foods, epidermoids, and venoms), gene therapy vectors, cellular therapy products, tissue engineered products for regenerative medicine, human tissue and cellular products used in transplantation, whole blood, plasma, and cellular blood products. The inactivated products include whole virion and subunit vaccines, allergenic extracts (such as molds), plasma derivatives, recombinant proteins, and blood components.
Adventitious agents are an important safety concern in biologics. Animal-derived raw materials are a potential source of virus contamination and can include cell substrates and biological fluids used for product manufacture as well as reagents used in cell culture growth, differentiation, and selection, such as serum, trypsin, antibodies, media/media components, antibiotics, cytokines, growth factors, collagenase, and DNase. The risk of adventitious virus contamination from source materials is greater in products that are not inactivated than in the case of inactivated products because inactivated products undergo virus inactivation and purification during downstream manufacturing. Therefore, the safety of products that are not inactivated relies primarily on the use of qualified raw materials, implementation of quality design concepts that are consistent with current good manufacturing practice (CGMP), and rigorous testing at multiple steps during manufacture.
History of Virus Contaminations Due to Raw Materials
Introduction of adventitious agents can occur at any step in production when cells are exposed to raw materials, and in many cases they may only be detected after amplification by growth in the cell substrate or in a bioreactor. There are many past and recent examples of viral contamination in different biological products due to raw materials, especially bovine serum and porcine trypsin. Some selected examples are shown in Table I. Contamination of Chinese hamster ovary (CHO) cells used for recombinant DNA products has been reported due to viruses in bovine serum, porcine trypsin, and possibly media components. Additionally, virus contamination may originate from the cell substrate due to the expression of endogenous infectious or defective retrovirus particles that are expressed from stably integrated sequences, which exist as a normal component of the host cell DNA—for example, rodent cell lines such as CHO cells (1) and mouse hybridoma cell lines used for monoclonal antibodies, and from chicken cell cultures used in viral vaccines (2–5)—or due to indigenous viruses that naturally occur in the species—for example, avian leukosis virus in embryonated hens eggs (6), simian virus type 40 (SV-40) in primary rhesus monkey kidney cell cultures (7, 8), and TCNL Alphanodavirus in a commercially available Trichoplusia ni–derived insect cell line, BTI-TN-5B1-4 (Tn5) (9). Several pathogenic viruses have been transmitted by blood and blood components (10–12). The need for continued vigilance and implementation of rigorous steps to deter introduction of adventitious viruses due to raw materials was demonstrated by recent reports of virus contamination such as calicivirus in a CHO cell–derived drug product (13) and by the discovery of porcine circovirus sequences in some Vero cell–derived viral vaccines (14, 15).
History of Virus Contamination Due to Raw Materialsa
Viral contamination due to raw materials can also influence adventitious agent testing results due to contamination of the target cells. In a recent example, the master cell bank was erroneously believed to contain an adventitious virus based upon 9CFR testing for bovine viruses (16); the equine rhinitis A virus (ERAV) contaminant was traced back to the use of nonqualified horse serum in the culture of Vero indicator cells used in the assay (17). These results highlight the need for rigorous management and use of qualified raw materials in test assays for evaluating the safety of biological products.
Safety Issues Related to Biological Raw Materials
Cell substrates are critical raw materials used in the manufacture of vaccines. Several different types of cell substrates are used for the production of U.S.-licensed viral vaccines (18–20). These include (1) animal tissues such as calf skin (lymph) for live viral vaccines such as smallpox vaccine (replaced by Vero cell–grown vaccine), mouse brain for Japanese encephalitis virus (JEV) vaccine (recently replaced by Vero cell–grown vaccine), embryonated chicken eggs for yellow fever vaccine, and influenza virus vaccines (live and inactivated); (2) primary cell cultures such as chicken embryo fibroblasts for live measles and mumps vaccines and for inactivated rabies vaccine; (3) diploid cells such as human WI-38 for live rubella vaccine and human MRC-5 for live varicella vaccine and for inactivated rabies, hepatitis A, and poliovirus vaccines; and (4) an African green monkey continuous cell line Vero for live rotavirus and smallpox vaccines and for inactivated poliovirus and recently JEV vaccine. Additionally, yeast cells are used for hepatitis B vaccine and for human papillomavirus vaccine, and most recently an insect cell line has been used for human papillomavirus vaccine.
As described earlier, adventitious agents are a major safety concern in cell substrates and may originate as (1) endogenous, genetically inherited retroviruses that exist as a normal part of the host cell DNA in the species of origin; (2) indigenous viruses occurring in the species of origin due to infection; or (3) viruses that were exogenously acquired due to cell culture passage by exposure in the laboratory to other cell lines or viruses, by use of nonqualified reagents, especially serum and trypsin, or due to handling and equipment (21–26). Adventitious agents may be introduced at many steps during product manufacture. This is best illustrated by a generic vaccine production scheme that indicates the steps generally used for development of the cell banks and the virus seeds for vaccine manufacture (Figure 1). The parental cell bank for the manufacture of a vaccine can be used directly, cloned, or undergo genetic modification in order to create a master cell bank from which a working cell bank may be derived. In the case of viral vaccines, the generation of the working virus seed, which is used to infect the cell bank for vaccine production, starts with initial isolation of the vaccine virus from tissue or cell culture. This is followed by the amplification of the virus in a cell substrate to create a master virus seed from which the working virus seed is derived. Thus, rigorous efforts should be made to minimize the risk of introducing adventitious agents during manufacture by developing a safety scheme that includes the use of qualified raw materials, the implementation of a program to manage the source reagents, and testing as specified by the regulatory authorities (16, 27–35).
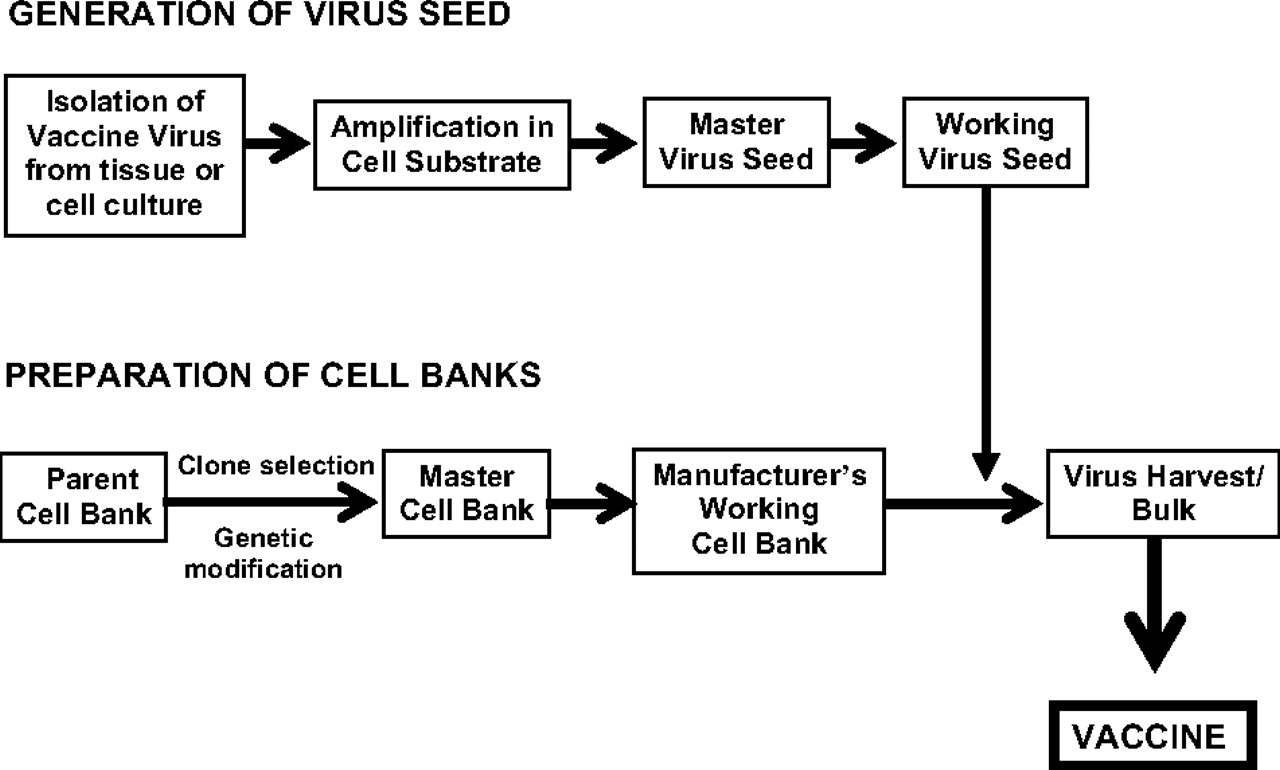
Generic vaccine production scheme.
Considerations for Developing a Comprehensive Testing Scheme for Product Safety
A comprehensive safety testing program can be established based upon assessment of the risk of cell substrate contamination and evaluation of risk reduction measures incorporated into the manufacturing process. The potential risk of cell substrate contamination can be assessed based on available information related to (1) the host species of origin (e.g., health of the donor, naturally occurring exogenous and endogenous viruses and specific exposure to other potential infectious agents such as animal vaccines); (2) cell substrate type and characteristics (e.g., primary, diploid, or continuous/immortalized cells and whether the phenotype is non-tumorigenic or tumorigenic); (3) the cell passage history (e.g., exposure in previous laboratories to other cell lines or viruses and use of nonqualified raw materials such as trypsin and serum); and (4) handling procedures and qualification of raw materials used during product manufacture. Evaluation of risk reduction includes (1) information regarding susceptibility of the cell substrates to known viruses, (2) presence of any virus clearance steps in the manufacturing process, and (3) use of qualified raw materials in production. Thus, based upon assessment of the risk of cell substrate contamination and evaluation of risk reduction, a comprehensive safety scheme can be developed that includes testing and use of qualified raw materials to reduce the risk of adventitious agent contamination in the final product.
Qualification and Management of Raw Materials
Qualification of raw materials includes documentation of the source, identity, purity, safety, and performance as desired or expected of a raw material. Thus, a qualification program should be implemented for the management of raw materials to assure identification and selection, suitability, characterization, and quality. Identification and selection of raw materials is based upon evaluation of suitability, toxicity, availability (consider alternate sources), consistency, contamination, and traceability (especially for human- and animal-derived raw materials (e.g., donor infectious disease status in case of humans and qualification and country of origin certification for herds providing fetal bovine serum during and after 1980). Suitability of use of raw materials in manufacturing is based upon the raw material's impact on safety, potency, and purity, which can be determined by assessment of potential risk of toxicity and adventitious agent contamination and removal if a large amount is used during manufacture. Characterization of raw materials should include testing for identity, purity, functionality, and safety (sterility, endotoxin, mycoplasma, and species-specific adventitious agents) (16, 32). Quality assurance of raw materials includes documentation of incoming receipts, segregation, inspection, and release of materials prior to use. This is achieved by vendor auditing and certification, Certificate of Analysis (COA) verification, testing or identity testing (as applicable), formal procedures and policies for out-of-specification materials, stability testing, and by archival sample storage.
It is also important to recognize which party is responsible for the control of various aspects of raw material usage. The source or supplier is responsible for assuring qualification of reagents and providing the COAs (36). However, the final responsibility of the quality of the components used in production is placed on the end user or manufacturer. The manufacturer is responsible for auditing the supplier and the testing labs and their results as well as for testing materials directly or during production. Inspection of the manufacturing facilities by regulatory authorities assures conformance with the regulations.
Efforts toward Designing Safety in Biologics
Efforts are being made toward minimizing the risks associated with biological raw materials, but these may need detailed consideration with regard to the specific product. (1) Replacements of animal-derived materials (e.g., serum-free cell cultures or use of non-animal-source materials such as plant-based or plant-derived) need evaluation of the cell phenotype characteristics and product yield; (2) replacements of primary cell cultures with well-characterized cell lines need evaluation of the product identity and potency particularly related to any mutations in the vaccine or vector virus or generation of novel recombinant viruses; (3) use of new technologies for virus inactivation and removal need validation of the effectiveness in the product-specific components; and (4) broad screening assays for adventitious agent detection and identification are emerging based upon novel technology platforms. These powerful research tools will provide a plethora of data that may need extensive interpretation and follow-up to assess biological significance and relevance of risk to humans.
The major goals of product manufacture are safety, high-quality product, and lot-to-lot consistency. The overall development of a strategy to assure product safety includes using qualified raw materials, control of the manufacturing process, and characterization of the final product consistent with relevant regulatory authorities and documents. Designing safety in viral vaccines is particularly challenging because contaminating agents need to be removed without significantly affecting the potency of the vaccine virus. This can be accomplished by (1) characterization of the cell substrate including evaluation of the cell phenotype (tumorigenicity); (2) qualification of the cell banks, virus seed, and biological raw materials including extensive testing of the seed and cell substrate and demonstration of the absence of detectable viruses by testing or providing COAs for the raw materials; (3) in-process testing to evaluate the bulk/production lots for known and novel viruses by a comprehensive testing plan; (4) process validation that may include designing an efficient process to avoid risk of contamination, to remove potential virus load, or to inactivate potential contaminating viruses; and (5) reduction of residual host cell materials in the final product by whole-cell removal and by cellular DNA and protein reduction. Furthermore, as new technologies and more sensitive assays can result in discovery of novel viruses, evaluation of potential risk to product safety may necessitate including additional assays designed for novel virus-specific detection. The establishment and implementation of a rigorous biocontainment and biovigilance safety plan is an important component of product manufacture that is essential to assure public health and confidence in biologics; this can be achieved by use of qualified raw materials that have been derived under concepts of quality design consistent with CGMP, by testing, as well as by the inclusion of steps for virus clearance in the manufacturing process.
Acknowledgments
I thank my colleagues for their contributions to the presentation: Robin Levis, Andrew Lewis, Nicolette DeVore, and Jay Slater in OVRR; Steven Oh in OCTGT; Indira Hewlett in OBRR; and Christopher Joneckis in the Office of the Director. I thank Robin Levis, Laraine Henchal, and Kathryn King for review of the manuscript.
- © PDA, Inc. 2010
References




{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- History of Virus Contaminations Due to Raw Materials
- Safety Issues Related to Biological Raw Materials
- Considerations for Developing a Comprehensive Testing Scheme for Product Safety
- Qualification and Management of Raw Materials
- Efforts toward Designing Safety in Biologics
- Acknowledgments
- References
- Figures & Data
- References
- Info & Metrics
Related Articles
Cited By...
- A Multicenter Study To Evaluate the Performance of High-Throughput Sequencing for Virus Detection
- Mouse Minute Virus (MMV) Contamination--A Case Study: Detection, Root Cause Determination, and Corrective Actions
- Detection of Latent Retroviruses in Vaccine-related Cell Substrates: Investigation of RT Activity Produced by Chemical Induction of Vero Cells
- Current Testing Methods and Challenges for Detection of Adventitious Viruses