
Abstract
With the publication of the quality guideline ICH Q9 “Quality Risk Management” by the International Conference on Harmonization, risk management has already become a standard requirement during the life cycle of a pharmaceutical product. Failure mode and effect analysis (FMEA) is a powerful risk analysis tool that has been used for decades in mechanical and electrical industries. However, the adaptation of the FMEA methodology to biopharmaceutical processes brings about some difficulties. The proposal presented here is intended to serve as a brief but nevertheless comprehensive and detailed guideline on how to conduct a biopharmaceutical process FMEA. It includes a detailed 1-to-10-scale FMEA rating table for occurrence, severity, and detectability of failures that has been especially designed for typical biopharmaceutical processes. The application for such a biopharmaceutical process FMEA is widespread. It can be useful whenever a biopharmaceutical manufacturing process is developed or scaled-up, or when it is transferred to a different manufacturing site. It may also be conducted during substantial optimization of an existing process or the development of a second-generation process. According to their resulting risk ratings, process parameters can be ranked for importance and important variables for process development, characterization, or validation can be identified.
LAY ABSTRACT: Health authorities around the world ask pharmaceutical companies to manage risk during development and manufacturing of pharmaceuticals. The so-called failure mode and effect analysis (FMEA) is an established risk analysis tool that has been used for decades in mechanical and electrical industries. However, the adaptation of the FMEA methodology to pharmaceutical processes that use modern biotechnology (biopharmaceutical processes) brings about some difficulties, because those biopharmaceutical processes differ from processes in mechanical and electrical industries. The proposal presented here explains how a biopharmaceutical process FMEA can be conducted. It includes a detailed 1-to-10-scale FMEA rating table for occurrence, severity, and detectability of failures that has been especially designed for typical biopharmaceutical processes. With the help of this guideline, different details of the manufacturing process can be ranked according to their potential risks, and this can help pharmaceutical companies to identify aspects with high potential risks and to react accordingly to improve the safety of medicines.
Introduction
Failure mode and effect analysis (FMEA) is a powerful quality management tool that results in a numerical ranking of risks aiding the prioritization of follow-up efforts. FMEA involves three orthogonal evaluations with ranges of 1 to 10 for each: severity (no effect to catastrophic), occurrence (never to always), and detectability (obvious to undetectable). A process FMEA can uncover process problems related to the manufacture of the product. Process FMEA has been used for decades in mechanical engineering and electrical industries (aerospace, automotive, and semiconductors) to optimize manufacturing operations. It can be easily adapted to the pharmaceutical industry when processes with a high number of batches that result in large amounts of identical parts are assessed, such as the manufacturing of tablets or medical devices. However, it is more difficult to apply to biopharmaceutical processes, because these processes are different. There is typically a low number of batches leading to large volumes of complex biochemical molecules in aqueous solutions.
Many books on the FMEA methodology focus on mechanical or electrical industries. A brief but excellent, nonindustry-specific introduction is given by McDermott, Mikulak, and Beauregard (1). There exist very few publications on biopharmaceutical process FMEA (2–7). Although these give helpful examples and present helpful case studies, they do not focus on adequate quantitative ranking details when used while actually performing FMEA. The proposal presented here is intended to serve as a brief but nevertheless comprehensive and detailed guideline on how to conduct a biopharmaceutical process FMEA. The centerpiece is a detailed, 1-to-10-scale FMEA rating table (Table I) especially created for biopharmaceutical processes. Interestingly, the severity rating is a general characteristic of the biopharmaceutical process itself, whereas both the occurrence and detectability ratings mainly depend on the manufacturing facility, staff, equipment, and systems applied. Therefore, a biopharmaceutical process FMEA as presented here can only be performed with a particular manufacturing facility in mind. However, the severity ratings can be determined independently of any facility.
Rating Guideline for Severity, Occurrence, and Detectability
The biopharmaceutical process FMEA approach described here is intended to be used as part of quality risk management according to the International Conference on Harmonization (ICH) quality guideline ICH Q9 (8) while considering the specific characteristics of biopharmaceutical manufacturing processes. Finally, the approach helps to broaden process understanding and therefore perfectly fits into the concept of quality by design (9).
Scope
A biopharmaceutical process FMEA is a useful tool whenever a biopharmaceutical manufacturing process is developed, scaled-up, or transferred to a different manufacturing site. It may also be conducted during substantial optimization of an existing process or the development of a second-generation process. During a biopharmaceutical process FMEA, process parameters can be evaluated for importance, and important variables for process development, characterization, or validation can be identified. The primary benefit of this tool is that it generates a ranked order of parameters that might require further development, characterization, or validation work.
A biopharmaceutical process FMEA requires time and resources. Because FMEA is team-based, several experts and specialists will have to be involved in the task. The basis of any FMEA is the input of the individual FMEA team members during discussions at team meetings.
While the organizational structure of operational functions within biopharmaceutical companies may differ, each production process can typically be divided into three sub-processes:
Upstream manufacturing of drug substance
Downstream manufacturing of drug substance
Manufacturing of drug product
An individual biopharmaceutical process FMEA can be conducted separately for each of the three sub-processes under the involvement of the subject matter experts from the respective operational functions to achieve optimal allocation of resources.
Team Members and Their Roles
Generally, the FMEA team size should be limited to enable efficient teamwork. Because FMEA requires extensive use of expert judgment, team members should represent all involved disciplines. These disciplines typically include but are not limited to the following groups:
Manufacturing and operations: upstream, downstream, or drug product (as applicable)
Science and development: upstream, downstream, or drug product (as applicable)
Analytical group(s) charged with quality control, in-process control, and process monitoring
Other group(s) charged with quality assurance, qualification and validation, engineering, regulatory, and project management
The following team roles are accomplished by individuals in addition to their function as normal FMEA team members:
FMEA Team LeaderThis person should be appointed as soon as the team is assembled. The team leader is responsible for setting up and facilitating meetings and for moderating the FMEA process. The team leader makes sure that the team is progressing toward the completion of the FMEA and aligns purpose, scope, and approach with the FMEA's of the other two sub-processes (as applicable).
Technical/Scientific Specialists
Technical/scientific specialists share their expertise and process understanding with the team to bring insight to the manufacturing process.
In addition to process specialists, other team members can also contribute to the FMEA. Additional technical/scientific experts, who are not team members, should be consulted whenever the team needs special pieces of information in order to make a decision during any FMEA team meeting.
Conducting a Biopharmaceutical Process FMEA
Prior to the start of any biopharmaceutical process FMEA, the purpose and scope of the FMEA as well as any deadlines should be clearly defined. In addition, the FMEA team leader and all team members should be appointed by management. The biopharmaceutical process FMEA can then be conducted according to the following sequential steps:
1. Review the Process
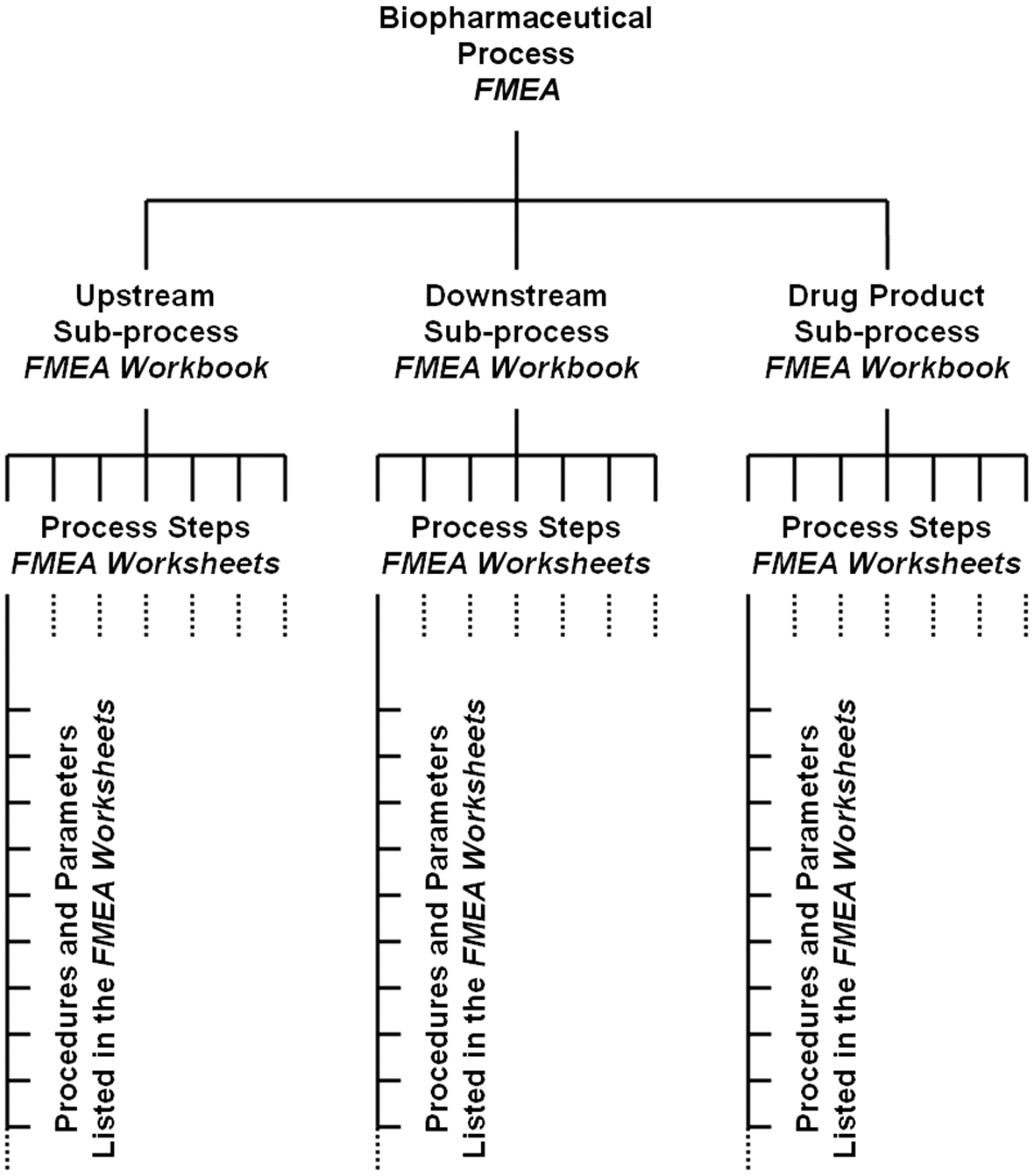
This should be finished prior to the first FMEA team meeting. An FMEA spreadsheet workbook should be prepared by the responsible upstream, downstream, or drug product manufacturing experts or by the responsible upstream, downstream, or drug product process development experts (as applicable). An upstream or downstream manufacturing sub-process should be divided into process steps. An individual worksheet (page) of the workbook should be used for each process step (unit operation). This hierarchical structure is visualized in Figure 1. The procedures and parameters as well as the corresponding target (set point), process ranges, and references to studies or literature (as applicable) of each process step should be entered in the respective worksheet in the order of process flow (see Figure 2 for an example template). The resulting workbook should then be distributed among the FMEA team. Each team member should review this workbook in preparation for the FMEA meetings.

A biopharmaceutical process can typically be divided into three sub-processes. For each sub-process, an individual FMEA workbook can be prepared, consisting of separate worksheets (pages), one for each process step (unit operation). The procedures performed during each process step and their corresponding process parameters can then be entered into the rows of the worksheets, following the process flow.

FMEA worksheet template proposing a minimum of required columns. Columns for additional information can be added as needed. One failure mode and its risk ratings are shown for the temperature of the production bioreactor unit operation (more details are described in the examples in the text). In reality, one unit operation will typically have several dozens of parameters listed.
Example: An upstream sub-process workbook is prepared by splitting the sub-process into its process steps (vial thaw and thaw culture, seed culture, scale-up bioreactors, production bioreactor, harvest).Each process step is then entered in an individual worksheet by listing all procedures and parameters in the order of process flow.
2. List Potential Failure Modes
This should be conducted during FMEA team meetings. For each procedure or parameter, potential failure modes are identified. If various failure modes can occur at a discrete procedure or parameter, there are two options. The failure modes can be combined in cases where they are similar to each other or trigger similar effects, or the corresponding procedure or parameter row in the spreadsheet can be copied. The decision whether failure modes should be grouped or not is made by the FMEA team individually for each potential group of failure modes.
Example: The parameter temperature can either be too high or too low, that is, there are two failure modes. If both failure modes result in the same failure effects, proceed in the FMEA. If high temperature triggers effects other than what low temperature does, copy the respective parameter row in the worksheet and consider risks for high and low temperature separately.
3. List Potential Effects of Each Failure Mode
This should be conducted during FMEA team meetings. If various failure effects can be triggered by a discrete failure mode, the corresponding failure mode row in the spreadsheet can be copied as needed.
Example: If the failure mode temperature too high can result in both reduced step yield and inferior product quality, it can be helpful to copy the failure mode row and assess the risks for both effects (reduced step yield and inferior product quality) separately (Figure 2).
4. Assign a Severity Rating (S) for Each Failure Effect (1 → 10)
This should be conducted during FMEA team meetings. Rate the severity of the failure effect but not the severity of any failure mode or cause. A high severity results in a high rating. Use Table I as a guideline. Biopharmaceutical products have a complex molecular structure, and the severity of potential changes of this molecular structure on product quality, efficacy, or safety needs to be judged by subject matter experts for each individual product. Hence, Table I can only provide a qualitative grading for potential effects on product quality, efficacy, or safety (severe, moderate, minor in severity ratings 8, 7, and 6, respectively). The final severity rating must be an integer from 1 to 10, which is based on the team's decisions.
Example: The failure mode temperature too high can result in both reduced step yield and inferior product quality, so the team decided to duplicate the failure mode row in the worksheet and rate both risks separately. If the team estimates that the reduced step yield would result in 20–50% loss of overall yield, then it can assign a severity of 6 for this failure effect. In addition, if a severe influence on product quality cannot be excluded, the team can end up with a severity rating of 8 for the second effect. Although the failure mode temperature too high can be caused by different things like a calibration error of the temperature sensor or a malfunction of the heating system, the team should not try to rate the severity of these failure causes (Figure 2). The occurrences of respective failure modes will be rated in the following step.
5. Assign an Occurrence Rating (O) for Each Failure Mode (1 → 10)
This should be conducted during FMEA team meetings. Enter potential causes for each failure mode in the worksheet to facilitate the rating. Rate the occurrence of the failure mode but not the occurrence of any failure effects. A high occurrence results in a high rating. Use Table I as a guideline. The table distinguishes between procedures and parameters that are monitored only once or a few times per batch and procedures and parameters that are monitored at frequent intervals or continuously. The first group of procedures and parameters is rated according to the number of occurrences of the failure mode (quantified by the percentage of batches in which the failure mode occurred or is expected to occur). The second group is rated according to the percentage of time at which the failure mode occurred or is expected to occur during an average process step. The final rating must be an integer from 1 to 10. Historical data, if available, should be evaluated to determine an exact rating. Otherwise, the rating has to be estimated based on the team's decisions.
Example: The failure mode temperature too high can result in both reduced step yield and inferior product quality. The team now has to rate the occurrence of a too-high temperature but not the occurrence of either reduced step yield or inferior product quality. As the step yield is checked at the end of the process step, the team has to find out what percent of batches are affected by this failure mode. However, if the temperature is monitored continuously during the process step, the team has to find out what percentage of the step duration is affected during a representative batch by this failure mode, because the continuous parameter monitoring could result in a higher frequency of minor excursions being detected during the processing of a batch.
6. Assign a Detectability Rating (D) for Each Failure Mode or Cause (10 → 1)
This should be conducted during FMEA team meetings. Enter current controls of the corresponding procedure or parameter in the worksheet to facilitate the rating. Rate the detectability of the failure mode or cause but not the detectability of any failure effects. A high detectability results in a low rating. Use Table I as a guideline. The final rating must be an integer from 1 to 10, which is based on the team's decisions.
Example: The failure mode temperature too high can result in both reduced step yield and inferior product quality. The team now has to rate the detectability of a too-high temperature and/or the detectability of its causes, such as a calibration error of the temperature sensor or a malfunction of the heating system. The team should not rate the detectability of either reduced step yield or inferior product quality.
7. Calculate the Risk Priority Number (RPN) for Each Effect
This should be conducted during FMEA team meetings. Multiply the severity (S), occurrence (O), and detectability (D) ratings in each row in the worksheet (RPN = O × S × D). The resulting risk priority number (RPN) values range from 1 to 1000.
Example: The team assigned a severity rating of 8 to the failure effect, an occurrence rating of 5 to the failure mode, and a detectability rating of 2 for the failure mode and its causes. This results in an RPN of 8 × 5 × 2 = 80 (Figure 2).
8. Prioritize the Failure Modes of Action
This should be conducted during FMEA team meetings. Those procedures or parameters resulting in high RPN values should then be subject to further considerations. A suitable RPN threshold for risk variables that are high (e.g., 100) is often difficult to predetermine because the RPN threshold depends on the assessed manufacturing process, the purpose and scope of the FMEA, and the distribution of the determined RPN values. A Pareto chart can facilitate the clustering of failure effects in categories of high risk and low risk (1, 4, 6, 9).
9. Risk Mitigation
This is only needed if it is applicable and within the scope of the FMEA. Take actions to eliminate or reduce the risks that are high. This should be conducted after or between FMEA team meetings by line functions that are not necessarily FMEA team members. Depending on the scope of the FMEA, examples for risk mitigation could be to perform a set of lab-scale process characterization studies to gain better process understanding or to implement additional automated alarm controls at risky unit operations.
10. Assign New S, O, and D and Calculate the New RPN after Risk Mitigation
This is only needed if it is applicable and within the scope of the FMEA. This should be conducted during FMEA team meetings. Refer to steps 4–7 listed above.
Conflict of Interest Declaration
The authors declare that they have no competing interests.
Acknowledgments
The authors wish to thank their colleagues from Boehringer Ingelheim at the biopharmaceutical site in Biberach, Germany, who were involved in various process FMEAs.
- © PDA, Inc. 2011




{kind=link}
{kind=link}