
Abstract
Bacterial penetration of integral sterilizing-grade 0.2 μm rated filters, although rare, is not a new phenomenon in the biopharmaceutical industry. It is recognized by both the Parenteral Drug Association and the Food and Drug Administration via recommended bacterial retention qualification (also commonly called filter validation) performed in relevant product fluids or suitable surrogates when necessary (1⇓–3). As noted in recent work, formulations such as some adjuvanted vaccines, liposome-based drug delivery solutions, and similar surfactant or emulsion-based product fluids increase the likelihood of such penetration events (4). Here we demonstrate that some 0.2 μm rated sterilizing-grade filters from different filter manufacturers may perform less effectively than expected when their membranes are challenged with one of these bacterial penetration risk-related solutions. Some filters provided very little sterility assurance (titer reductions < 6 logs) and others provided substantial sterility assurance (titer reduction > 8 log). From this, it is clear that the product formulation most likely to lead to filter penetration must be identified early in the design process to facilitate process design and minimize qualification costs. In this way, the solution can be matched with the appropriate sterilizing-grade filter and the appropriate process conditions.
LAY ABSTRACT: Bacterial penetration of intact sterilizing-grade filters during filter qualification, although rare, is not a new phenomenon in the biopharmaceutical industry. Because these incidences are identified in the filter validation process, there is no risk to the drug product end-user, but these failures do incur additional expense to the pharmaceutical manufacturer and could prevent the manufacture of very important drug formulations. Formulations such as some adjuvanted vaccines, liposome-based drug delivery solutions, and similar surfactant or emulsion-based product fluids have been documented to lead to an increased risk of penetration events. Here we demonstrate that some sterilizing-grade filters may perform differently from expected when their membranes are challenged with one of these solutions related to bacterial penetration risk. Some filters provided very little sterility assurance and others provided substantial sterility assurance. From this, it is clear that the pharmaceutical products and product formulation most likely to lead to filter penetration must be identified early in the design process to facilitate process design and minimize qualification costs. Moreover, that solution should be matched with the appropriate sterilizing-grade filter and process conditions to ensure expected sterility.
- Liposome solution
- Emulsions
- Sterile filtration
- Vaccines
- Titer reduction
- Drug delivery
- Bacterial penetration
Introduction
Use of particulate-like drug delivery systems for targeted drug delivery is becoming increasingly important (5, 6). Ideally, targeted drug delivery enhances precise delivery of a pharmaceutical agent, prevents premature metabolic breakdown of the pharmaceutical, and decreases the occurrence or severity of toxic side effects. In doing so, it protects both the patient and the drug and may even permit the delivery of pharmaceuticals that might otherwise be essentially unavailable to the patient (especially pharmaceuticals that are poorly water-soluble or extremely toxic). They can be designed to reach bacteria hidden in protected niches (7) (which are otherwise resistant to antibiotics), penetrate the skin for transdermal drug delivery, or protect pH-sensitive pharmaceuticals as they are transported through the gastrointestinal system (8). These particulate-like carriers are being developed for (or currently used for) delivery of chemotherapeutics, HIV/AIDS drugs, colonic drugs, antibiotics, and others (5, 8).
The drug delivery systems described here are frequently found as liposome solutions or as emulsions. Liposome solutions are composed of artificial phospholipid vesicles with a lipid bilayer encasing the pharmaceutical. Emulsions take similar vesicle forms but generally are surrounded by a lipid monolayer protecting a hydrophobic core in which the drug is encapsulated. The external surface of the liposome or vesicle is useful for targeted drug delivery because these surfaces can be modified to enhance vesicle attachment to targeted sites.
In delivering drugs with these particulate carriers, two important obstacles to effective and safe drug delivery are minimized, resulting in protection of both the patient and the pharmaceutical, but a new challenge has arisen: ensuring successful sterilizing filtration of the resulting suspension. In previously published work, an analysis of instances of bacterial penetration through various sterilizing-grade filters indicated that these particulate drug delivery suspensions were found to be at a greater risk of bacterial penetration than nearly all other solutions (4). Although the presence of lipids and a low fluid surface tension was also associated with penetration, liposome and related solutions were the most likely test solutions to result in a penetrative event. Given the increasing interest in use of these solutions as a vehicle for pharmaceutical delivery, it becomes clear that effective recommendations for successful sterile filtration of these solutions are needed.
While organisms smaller than Brevundimonas diminuta such as Hydrogenophaga pseudoflava and Ralstonia pickettii in nutrient-deprived fluids have been shown to consistently penetrate sterilizing-grade filters, B. diminuta penetration of sterilizing-grade filters is very rarely detected (9). Further, all sterilizing-grade filters are validated prior to use with a filter validation test, so there is no risk to the final drug product user because the rare instances where bacterial penetration occurs is detected prior to actual pharmaceutical drug product production (3). However, a failure of filter qualification during what is commonly called the “filter validation stage” is costly to pharmaceutical drug manufacturing.
In sterilizing filtration qualification, it is necessary to demonstrate effective bacterial retention by the filter membrane under the conditions of actual use (1⇓–3). One aspect of this qualification includes a bacterial retention test with B. diminuta (and, where applicable, any suspect process isolate) under user-defined, worst-case conditions. This bacterial challenge must demonstrate complete retention of the appropriate challenge organism(s) by the test filter membrane when challenged with a minimum of 1 × 107 colony forming units (CFU) per square centimeter of effective filter area (EFA) (i.e., CFU/cm2). This is an exceedingly high challenge load, well above the expected bioburden of a drug manufacturing process. As such, it provides a very robust test of a sterilizing-grade filter and a large safety factor.
While 0.2 (or 0.22) μm rated filters are the accepted industry standard for sterilizing-grade filtration, the rare penetration occurrence creates an opportunity in the industry to both reduce the likelihood of such events during filter validation testing and begin taking steps to shed light on the mechanisms at work. Fortunately, due to the high microbial load and worst-case process parameter nature of sterilizing filtration validation bacterial challenges, penetration events are determined and characterized well before patient and commercial risk is possible, so there is no risk of contaminated product reaching the end user. The risk, in this case, is to the pharmaceutical manufacturer with regard to the cost of multiple filter validations to determine the correct manufacturing process parameters and filter for the particular product. If a bacterial challenge fails, then the validation study is usually repeated with a new filter and/or with new user-defined parameters to ensure the final process provides adequate sterility assurance. However, retesting is costly for both the filtration validation service provider and the drug or vaccine manufacturer. These costs include delays to market as well as the financial impact of materials required for multiple retests.
Materials and Methods
Preparation of Cultures and Challenge Suspensions
A B. diminuta ATCC No. 19146 challenge suspension was prepared from cells previously grown to stationary phase in large-scale batch cultures and stored as a frozen cell paste until used (10). The thawed cells were suspended in liposome solution at a cell density of approximately 106 CFU/mL, which was then metered (at approximately 1 mL/min) into the recirculating liposome solution.
Challenge Solution
The selected liposome solution was a neutral, 10% cholesterol-based solution prepared by an independent laboratory. Prior to use in the bacterial challenge, the liposome solution was placed in an ultrasonic cleaning bath, cavitated for 5 min, and pre-filtered with a 1.25 μm rated filter to remove any remaining large vesicle aggregates.
Test Filters
All test filters were commercially available filters from four different manufacturers, 0.2 (or 0.22) μm rated sterilizing-grade filters, with the exception of one filter that was 0.1 μm rated, also commercially available. Not all filters were commercially available as 47 mm discs. Those filters that were not obtainable as 47 mm discs were purchased as small capsules with a nearly identical surface area. All the data was normalized per unit filter area (cm2) where appropriate (see Table I). Filters A through E were composed of polyethersulfone (PES). Filters F and G were composed of both PES and polyvinylidene fluoride (PVDF) layers.
Results of B. diminuta Bacterial Challenge Tests of Various Commercially Available 0.2 μm Rated Sterilizing-Grade Filters and One 0.1 μm Rated Sterilizing-Grade Filter Using a Liposome Solution as the Challenge Solution
Equipment Preparation
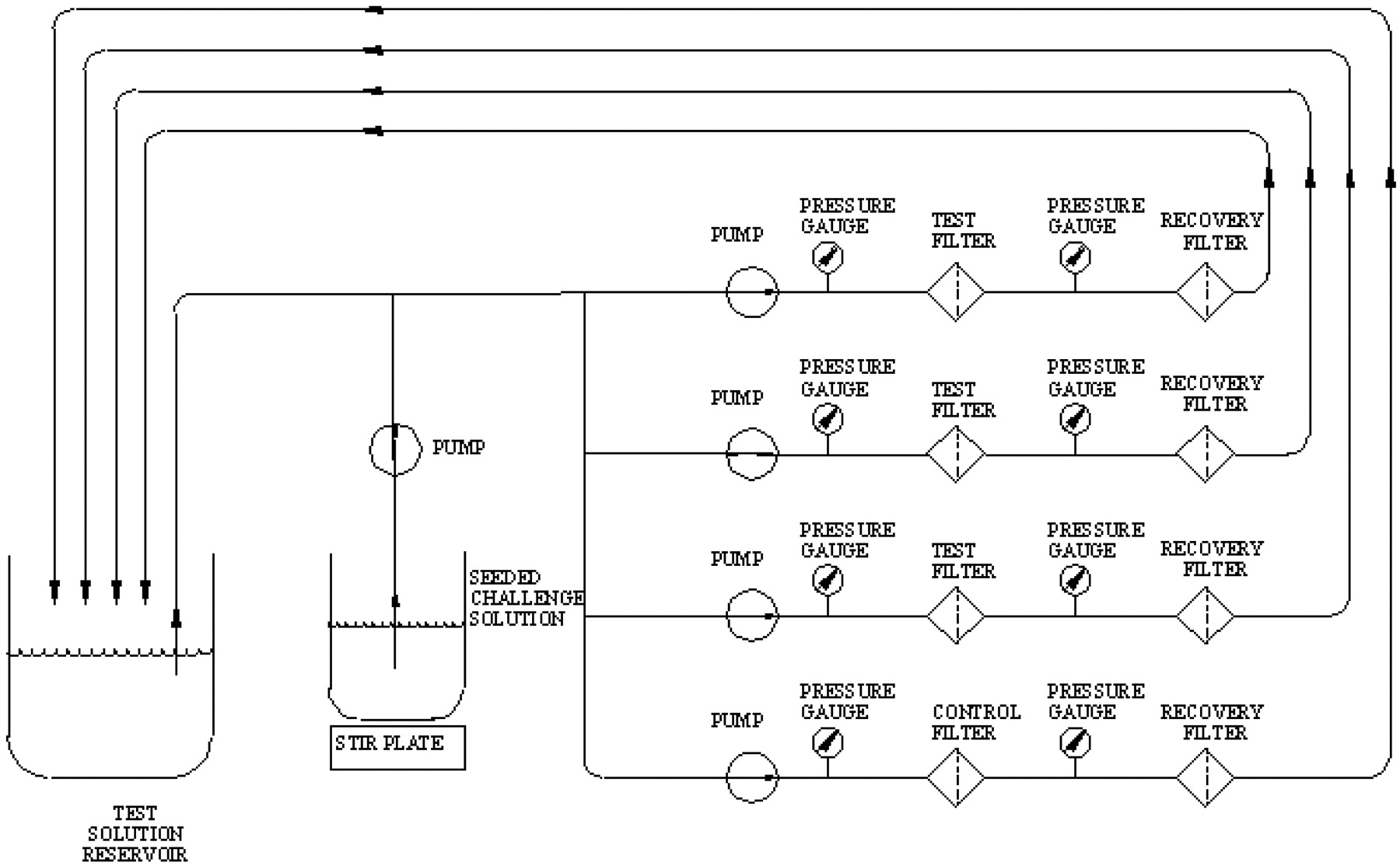
All the test filters were either challenged in capsule assemblies (as purchased) or as 47 mm discs mounted in an appropriate housing. In all cases, three test filters were tested in parallel simultaneously with one penetration control filter. The penetration control filter consisted of a 0.45 μm rated Pall Ultipor® N66 filter (P/N FTKNXG) and was used to provide assurance that the test organism was sufficiently dispersed and small enough to penetrate 0.45 μm rated membranes. Recovery filter discs (0.2 μm rated Ultipor N66-P/N NR047100) were installed downstream of the test filters (see Figure 1). All of the effluent was passed through the recovery filters.

Schematic of the filtration setup.
To confirm the pore size rating and to evaluate the integrity of the filter, each test filter was subjected to an installation bubble point test and the resulting data was evaluated as described by the manufacturer. All passed acceptance criteria. The entire apparatus, including the downstream tubing, was sterilized by autoclaving at 121 °C for 50 min.
Challenge Delivery Operating Parameters
The liposome solution was briefly recirculated through the test and control filters for a minimum of 5 min. Once stable recirculation of the liposome solution was established, B. diminuta, also suspended in liposome solution, was injected into the recirculating liposome solution at 1 mL/min for 120 min. The challenge was run at room temperature and monitored with a thermometer to stay within 20–25 °C. The challenge was limited to 120 min for ease of testing and due to the fact that penetration was detected within the first 120 min.
Throughout the challenge, differential pressure was maintained by means of a pump. Pressure differentials across the test filters were monitored by means of pressure gauges installed upstream and downstream of the test housings. Flow rates were monitored and adjusted to equalize the differential pressure conditions across the various test filters, as much as possible, although slightly elevated pressure was required to complete the test using filters E, F, and G. The average flow rate and differential pressure over the course of the test, for each filter, is reported in Table I.
At the completion of the challenge, the recovery filter discs were flushed with 2000 mL sterile deionized (DI) water, removed from the housings, and plated on trypticase soy agar (TSA) plates.
A sample was removed from the challenge suspension reservoir upstream of the test filters so as to determine the titer of the challenge pre- and post-challenge. A sample of the influent challenge suspension was also microscopically evaluated for cell dispersion (using the acceptance criteria of 80% monodispersion) and was determined post-challenge. All plates were incubated at 30 ± 2 °C for 7 days. The test filters were also subjected to post-challenge bubble point tests.
Results and Discussion
The results of B. diminuta bacterial challenge tests of various commercially available 0.2 (or 0.22) μm rated sterilizing-grade filters and one 0.1 μm rated sterilizing-grade filter, using a liposome solution as the challenge solution, can be found in Table I. All filters were subjected to similar differential pressures and nearly identical challenge concentrations (per cm2 EFA).
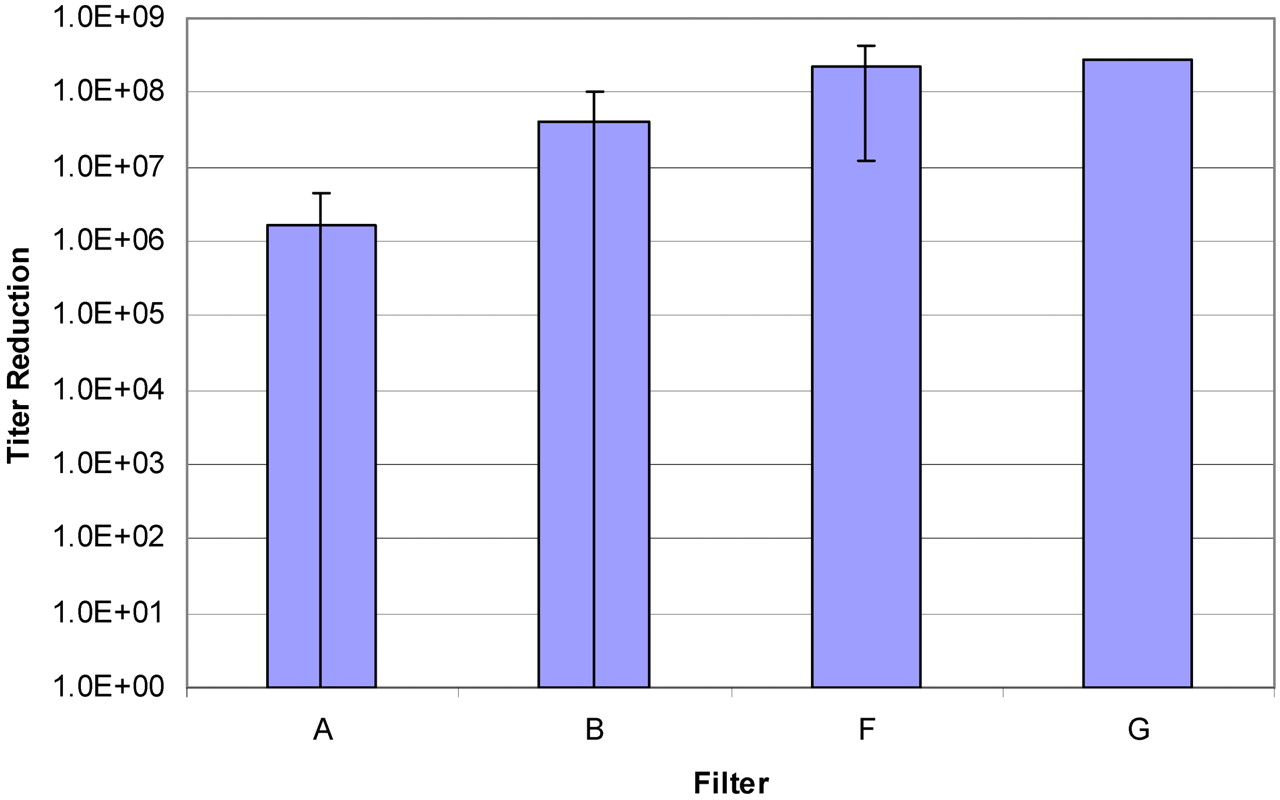
Five filters (including a Pall membrane and four competitor filters) of a total of seven tested filter types resulted in penetration in all tests (Table I). Where possible to calculate, the titer reductions varied from less than 106 (the minimum titer reduction detected in these tests) to greater than 108 (Figure 2). All 0.2 μm rated filters showed some degree of penetration; only the 0.1 μm rated filter (G) showed 100% retention, and one 0.2 μm rated filter showed a consistently high titer reduction (F), meaning it provided a higher degree of sterility assurance (when compared to the other tested 0.2 μm rated filters) under the conditions of this test.

A comparison of the average titer reduction detected for Filters A, B, F, and G. Filters C, D, and E were not included because the titer reduction for those filters was below the detection limit in all cases (effectively 0). In the case of Filter A, to calculate the average titer reduction for that filter, a value of 0 was used when the titer reduction was below the detection limit (two instances).
Given the lower number of filters showing penetration, Filter F was retested to confirm the observation, showing complete retention in three of six trials. Thus, of the 0.2 μm rated filters tested, the likelihood of bacterial penetration with the liposome carrier fluid used in this bacterial challenge study was greatly reduced with Filter F, the sterilizing-grade filter (an asymmetric 0.2 μm upstream layer over a symmetric 0.2 μm layer). Bacterial penetration was prevented with Filter G, the 0.1 μm rated filter (an asymmetric 0.2 μm layer over symmetric 0.1 μm layers).
Fluid flux is a measure of the rate of fluid flow through the filter per unit area of the filter (mL/min/cm2). Maximum fluid flux is frequently used as the preferred selection criteria when choosing a filter for a manufacturing process. High fluid flux is generally preferred due to the decreased processing time provided. It is a logical choice, and choosing a filter that provides the maximum fluid flux potentially reduces manufacturing costs. However, from this data, it is also evident that maximized fluid flux may not be the most effective filter selection criteria when filtering these particular fluids. Some loss in flux may be necessary in the interest of increasing sterility assurance. Filter D, for example, provided a high flux (0.8 mL/min/cm2) but poor sterility assurance (100% penetration), and filter G provided a much lower flux (0.18 mL/min/cm2) but a very high degree of sterility assurance (100% retention).
In addition to fluid flux, the effect of the filtration on the pharmaceutical drug product or product formulation is a legitimate concern. The end product must be sterile, and further, it must remain unaltered. Although there was little loss in flux between Filters G and F, it is possible that some characteristics of the liposome solution (such as size distribution of the micelles) may have been altered after passing through the 0.1 μm rated membrane compared to the 0.2 μm rated filter. If the drug product was altered in a way that affects drug efficacy, then the use of the filter with a 0.1 m rating would not suffice even if it provided a sterile effluent. It should be noted that, in the testing described here, analysis of the filter effluent other than for bioburden content (the test organism only) was outside the scope of this study, but the effect of the filtration on the drug product would need evaluation in actual use, when designing the manufacturing process.
Consequently, it is necessary for the user to plan carefully when filtering these suspensions that are at high risk for bacterial penetration. It is also important that this planning and filter membrane qualification occur early in the manufacturing process design, so that adequate sterility assurance is reached in the end, and sterilizing filtration validation proceeds without the need for re-testing or modifications to the manufacturing process. This will require careful consideration of the process so that quality (bacterial content as well as product efficacy/yield after filtration) and product integrity are designed into the process from the beginning.
In some cases, it may be difficult or not possible to meet the demands of a worst-case filter qualification (testing with an artificially high bacterial load) with these solutions. In such cases, if the potential medical benefit provided by the drug or drug formulation is greater than the risk incurred, then it may be necessary to consider the actual bioburden level of the specific process and assure sterility with respect to the actual process. To do so it may be necessary to generate the data to justify such an approach to regulatory bodies. This flexibility is necessary to balance the need for sterility assurance with the need to produce a vitally important product. These situations may be best initially handled on a case by case basis.
In summary, successful sterilizing filtration validation, when using liposome and closely related solutions at high risk for bacterial penetration, is a balance between robust filter qualification and product yield, and therefore requires careful consideration of the specific characteristics of (1) the process solution, (2) the filter chosen, (3) the desired end product, and (4) the desired process parameters, all to ultimately ensure sterility of the final product. Most importantly, all these elements must be balanced early in the process design, that is, in the initial filter selection and qualification prior to the final validation study.
Conflict of Interest Statement
Both authors work for Pall Corporation.
- © PDA, Inc. 2012




{kind=link}
{kind=link}