
Abstract
Interest in human factors usability testing has seen a sharp increase from manufacturers bringing new injectable pharmaceutical combination products to market and from regulators reviewing and approving submissions. This paper highlights the special regulatory considerations in the planning, execution, and reporting of human factor usability studies for injectable combination products. The paper describes recent human factors examples that capture and convey important sponsor learning for drug/device combination products. Special emphasis is placed on the recent focus across U.S. Food and Drug Administration centers, offices, and divisions in issuing new draft guidance outlining expectations in the execution and reporting of usability testing. Insight is provided into how the new guidance has been put into practice in the development and review of injectable combination products, and some of the unwritten recommendations/expectations that have been gleaned from these regulatory interactions are identified. The paper also describes future areas of opportunity for regulatory guidance based on reflections from over two dozen recent combination product human factor studies covering from early design development and testing through to the reporting of human factors results in the final submission.
LAY ABSTRACT: Human factors is the study of the interaction of people and technology to ensure the safety and effectiveness of that interaction and to improve human/device compatibility, including the user interface, instructions, and training programs to avoid use error. The enhanced focus on human factors usability assessments for injectable combination products is an acknowledgement by regulators and industry that the device mechanics are typically quite reliable and that device risk hazards are likely due to device usability. Use errors can occur when the device is not being used as intended or the design features are less than optimal. Human factors testing, analysis, and validation helps to identify and eliminate use errors by informing appropriate mitigation strategies to ensure the device design provides the optimum use of injectable drug/device combination products.
Introduction
In recent years there has been a sharp rise in pharmaceutical products that must be administered subcutaneously or intramuscularly due to the explosion in the development of protein therapeutics and monoclonal antibodies. This has resulted in a corresponding increase in the administration of these products through injectable drug/device combination products. Combination products have the potential to make the delivery of pharmaceuticals safer, more effective, and more acceptable to patients, but these products can also raise complex scientific and technical development issues. The drug/device combination product must be thought of as a single product. Patients consider the drug they take and the device that delivers it as inseparable—equal partners in the patient experience. The drug helps in the treatment or cure of disease, while a well-designed device can have a number of positive effects that are both clinical and emotional. The device can touch upon issues related to acceptance, initiation, comfort, convenience, and even treatment compliance, all leading to better patient outcomes. However, the incorporation of a device can come with added risk because the device portion of the product adds mechanical complexity and usability considerations. The drug portion of the combination product may have demonstrated beneficial efficacy and acceptable benefit/risk, but the full utility of the drug cannot be realized if the device portion is less than optimal in its delivery of the drug.
Human factors (HF) is the study of the interaction of people and technology to ensure the safety and effectiveness of that interaction to improve human/device compatibility, including the use interface, user instructions, and training programs to avoid user error. It is important to ensure that the device user interface is optimized, with potential unforeseen use errors identified and eliminated (or at least reduced to a minimum) through appropriate mitigation (e.g., device design interactions, lockouts, training, instructions for use) (1). Formative HF studies have been critical to ensure proper injectable combination product design and have worked best through the systematic consideration of HF studies in the context of development of the device user interface. HF testing, analysis, and validation are crucial to ensure the device design provides the optimum use of the drug/device combination product.
Renewed Emphasis on HF
There has been a significant uptick in interest among regulators and industry in the planning and execution of HF and usability testing for injectable products. HF engineering has played a valuable role in identifying, evaluating, and addressing use-related hazards during device design development (2). HF studies to optimize device design and labeling have positively affected safety and effectiveness and have also reduced the potential of product recalls, user complaints, product liability claims, and need for post-approval modifications. With the appropriate HF study evaluations and mitigation responses, the increased usability and product appeal (e.g., “fits well in my hand”) can even be a competitive advantage in a crowded drug/device market.
The two main HF international standards: IEC 62366, Medical Devices—Application of Usability Engineering to Medical Devices (2007) (3) and ANSI/AAMI HE75, Human Factors Engineering—Design of Medical Devices (2009) (4) provide detailed understanding of the design considerations and application of usability testing that can be applied to injectable combination products. The U. S. Food and Drug Administration (FDA) also recognizes ISO 14971-1:2007 Medical Devices—Application of Risk Management to Medical Devices (5) and IEC 60601-1-8:2006 Medical Electrical Equipment – Part 1-8: General Requirements for Safety-Collateral Standard Alarm Systems-Requirements, Tests and Guidance—General Requirements and Guidelines for Alarm Systems in Medical Equipment (General) (6) that should be followed to conduct appropriate HF/usability methods for device design development and assessment. A fifth standard describing HF process is AAMI/ANSI HE74:2001/(R)2009, Human Factors Design Process for Medical Devices (7).
Regulators continue to work with manufacturers to address concerns that preventable dispensing errors, medication differentiation, and/or adverse events related to use errors can be further reduced by ensuring adequate HF studies are performed. These concerns have led the FDA to raise awareness of the importance of usability testing and to clarify its position on HF engineering expectations (8–16). A key HF regulatory guidance is the relatively new June 2011 FDA Draft Guidance for Industry and Food and Drug Administration Staff: Applying Human Factors and Usability Engineering to Optimize Medical Device Design (9). The draft HF guidance does a very good job describing how HF methods can be applied during device design to effectively manage use-related risks. The draft HF guidance also provides explicit directions on the level of detail of HF information to be summarized and reported in the submission.
HF study results (previously known in-house as use studies) had always been kept internally on file as technical reports. There was an apparent shift in FDA expectation that the HF results were to be submitted in the New Drug Application/Biological License Application (NDA/BLA). In fact, the FDA rejected a combination product submission due to lack of HF study results information. This resulted in the refinement of (1) the HF study plans and execution, (2) the regulatory interactions to finalize the HF study protocols, and (3) the inclusion of the summary HF engineering/usability engineering (HFE/UE) report in the main submission (with the summative HF technical report often included in the submission appendix).
The enhanced focus on usability assessments is an acknowledgement by regulators and industry that failures are most likely not due to mechanical failure of the device but rather due to device usability. The device's mechanics work just fine, but use errors occur when the device is not being used as intended. This suggests that injection devices may not be as intuitive as product sponsors envisioned. People hold the device in a certain orientation or take use steps that were not intended, which makes design features less than optimal. For example, in the design of a dose button on an injector, if the button looks too much like a knob (as in a doorknob or the knob on a radio), then the user is inclined to twist or turn it. Therefore, it's important to avoid the appearance of a knob and to design physical features unique to a button to be pushed down. Formative HF studies informed design modifications that led to a scalloped top of the button, so it looks nothing like a knob and has the appearance and feel to fit the contours of a person's thumb.
Scope of HF
HF includes the “user interface”, which is defined broadly to include all components of a device with which the user interacts, the size and configuration of the device (particularly as these injectable combination products are hand-held devices), and controls and displays (i.e., those parts of the device that users see, touch, and hear). The user interface also includes the device labeling, which includes package labels, carton, instructions in user manuals, package inserts, instructions on the device itself, quick reference guides, and any accompanying informational materials.
The most effective strategy to address use-related errors in early-phase combination product development is to focus on improvements to the design of the device user interface. To the greatest extent possible, the user interface should convey the concept for correct operation through its appearance and operation (“look and feel”). Asking questions on, “How intuitive is the use of the device?” and “Will it be natural for the patient to use the device in a safe and effective manner?” help to gain insight into how the device will be used.
What has been found in over two dozen formative evaluation HF studies, especially when observing users who received no prior training on injectable combination products—and even more so when users decided to just discard the carton and instructions for use (IFU) and “go it alone”—was a myriad of creative and unforeseen ways to use the product. Users tend to expect devices and device components to operate in ways that are consistent with their experience with other similar devices, user interface elements, or perceived device output. For example, when users were asked why they did not wait the full allotted time as described in the IFU to hold the device to the skin for the injection to be completed, users often said they thought the injection would be instantaneous. More than once a user would cite the “less than a second” injections like they remembered seeing on science fiction shows (e.g., Star Trek TV show).
Addressing use-related hazards by modifying the device design is generally more effective than revising the labeling or training. Labeling might not be accessible when needed, and training (if provided) depends on memory, which might not be complete or accurate.
Risk Evaluation and Mitigation
The process of categorizing the tasks required to operate the injectable combination product begins with the risk management process. This process uses a cross-functional team whose members are familiar with the drug component of the product and disease indication to consider the possible hazards that might occur from the use of, or failure of, the device component. Examples of the hazards identified for injectable combination products include underdoses, overdoses, and incorrect injection site. Each of the potential hazards identified were assigned a severity ranking, using the scale shown in Table I.
Hazard Severity Rankings
Risk analysis is performed to determine the chain of events that may result in exposure of the patient or other user to the hazards identified in the hazard analysis process. The application failure modes and effects analysis (AFMEA) risk analysis tool is used to identify use errors that might occur at each device operation step and the potential harm associated with each use error. Based on this AFMEA evaluation, the tasks required to achieve the primary function of the device constituent of the combination product (i.e., to deliver a subcutaneous injection of medication) were categorized according to the following criteria (5):
Critical Task/Use Step—a task that could potentially result in clinically significant harm if omitted or performed incorrectly. These are tasks with potential use errors that have a Severity Ranking of 4 (Major) or 5 (Severe).
Essential Task/Use Step—a task that if omitted or not performed correctly could potentially result in failure to achieve the primary function of the device constituent of the combination product. These are tasks with Severity Rankings 1 through 3 and they must be performed in order to deliver the injection.
Based on FDA feedback, other tasks that are not essential or critical in the IFU and do not directly contribute to the completion of the primary function of the device constituent of the product are not listed in the HF study protocol. These are tasks like washing hands or pressing cotton ball over the injection site (after removing needle from skin). These tasks have been called ancillary, and the moderators take note and document if the study subject does or does not perform each of these steps.
The HF studies are critical in obtaining a complete understanding of how the injectable combination product will be used so as to gain a full assessment of potential use errors. All known use errors are incorporated in the risk analysis for the delivery system. The use errors are taken into account when selecting and reassessing the critical and essential tasks to be evaluated as part of the HF analyses. The same thorough evaluation goes for new devices in development or revisions to a marketed device (e.g., continuous improvement iterations) for the HF analysis, including validation testing, to assure any design modifications have properly mitigated risks associated with a use error (13).
Risk mitigation strategies employed in the development of injectable combination products include modifying the interface design, user instructions, and/or training to address potential use errors and/or to optimize the user interface. If design flaws that could have negative clinical impact on patients are identified, then these flaws need to be address through further design modifications pre-submission. Planning to address design flaws in subsequent versions of the device, rather than immediate mitigation prior to the initial submission, is not acceptable. Regardless of the strategy used, subsequent testing is important to ensure modifications/interventions have successfully controlled the use-related hazards and that these risk mitigation efforts have not inadvertently introduced any new risks. The analysis of residual risk should determine if design modifications would further reduce use errors, and if not, then the analysis needs to demonstrate the impossibility or impracticality of reducing these risks further. The analysis needs to clearly state that any remaining residual risk is outweighed by the advantages offered by the drug/device combination product.
The evaluation and assessment of the success of the mitigation strategies at reducing risks to acceptable levels, either by reducing the probability of the occurrence of the problem or by reducing the severity of the potential consequence of the problem, continues to be a great source of discussion with manufacturers and regulators. The residual risk has been acceptable if it is reasonably limited, extremely difficult to eliminate or further reduce (with the potential of creating new risks), and outweighed by the device's benefits. Formative HF studies have been run where the use errors were reduced to very low levels but were not completely eliminated. The summative HF protocol was submitted to the FDA with the rationale for why the use errors were eliminated down to their lowest practical level. Several questions were asked of the FDA requesting guidance on the acceptability of not having eliminated a specific use error down to zero occurrences. The FDA agreed that use errors identified had been appropriately mitigated through design modifications and instructions, and the FDA acknowledged that in some cases residual use errors remain and cannot be reduced to zero occurrences. The FDA determined the specific use error had been reduced down to reasonable levels in the to-be-marketed device.
In the end, the sponsor needs to have the confidence to conclude in the HFE/UE report that the product has been found to be reasonably safe and effective for the intended users, uses, and use environments. Lilly states verbatim the wording of the June 2011 draft HF guidance of “any residual risk that remains after the validation testing would not be further reduced by modification of design of the user interface (including any accessories and the IFU) and is outweighed by the benefits that may be derived from the device's use”. Knowing that this is the statement included in the submission drives the internal discussions in evaluating the acceptability of submitting the drug/device combination product to the regulators for marketing approval.
Formative Evaluations and Summative Validation HF Testing
The subject of HFE/UE is understanding and optimizing how people interact with technology, and as it is applied to injectables, how people interact with the drug/device combination product. The formative and summative (validation) testing is used to demonstrate that the potential use errors have been eliminated or minimized to reasonable levels when run under realistic conditions. The typical HF engineering process for medical devices takes the following steps:
Gather historical information—market research studies, user complaints, adverse event data.
Determine the intended user profile (also known as user groups) and use environment.
Complete the initial risk analysis incorporating risk controls into the product design.
Conduct the formative HF studies and update the risk analysis and change the product design if needed.
Conduct another formative HF study to confirm that risks are adequately mitigated.
Conduct the summative (design validation) HF study. Include a HF summary in the submission summarizing the formative studies, the summative studies, and the risk analysis.
Formative use studies provide an effective way to assess device designs and labeling (especially Instructions for Use). During formative HF studies, use errors are evaluated leading to design modifications to eliminate or control use-related hazards. Table II shows an example of use problems broken down into use errors, close calls, or difficulties for critical and essential use steps in a formative HF study. These results are used to develop mitigation strategies that include design modifications, enhanced instructions, and/or training to further reduce the probability of encountering use problems in subsequent HF testing.
Example of Observed Use Problems by Critical/Essential Use Steps
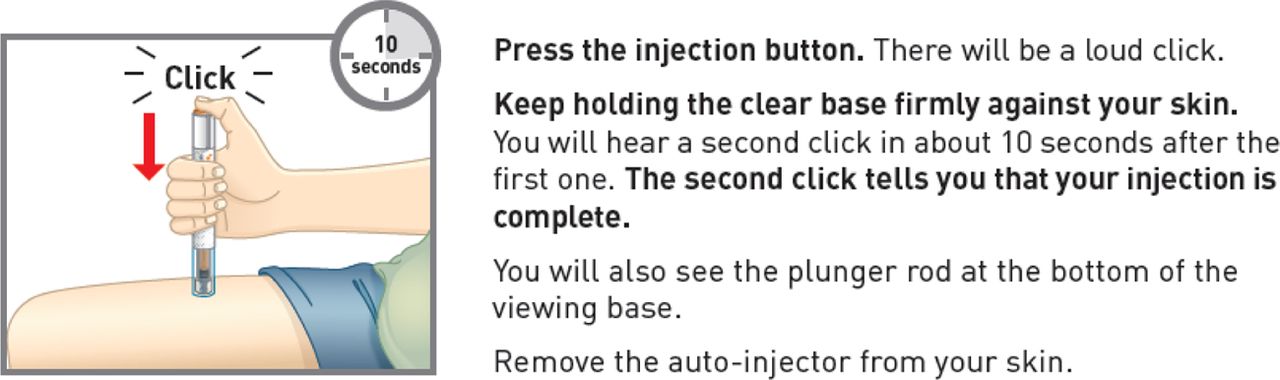
An example of enhanced instructions is to address the reoccurring use error for injectable products of early lift-off, an action where the user did not hold the device to the injection site for the full timing or auditory prompt. Patients had trouble distinguishing auditory signals when being instructed to not remove the device from the skin until a click was heard. The FDA recommended that a time-based instruction would be a better way to ensure that the full dose is delivered. The FDA stated that it is difficult to predict if patient would recognize the difference between a soft click versus a loud click and may remove the device before injecting the full dose. However, formative HF studies showed that patients have trouble counting in a consistent manner, and again removing the device from the skin is a concern.
The HF studies demonstrated that actually a combination of both timing and auditory prompts provides the best instruction to the patients to ensure the full dose is delivered. Figure 1 shows an example of a step in the IFU that combines the auditory and timing prompts for task completion. The revised IFU was tested in formative HF study and resulted in a sharp increase in the user group's success rates, especially with elderly patients and caregivers, in their ability to administer the full injection dose.

Example of Instructions for Use statements and graphics providing both timing and auditory prompts to ensure task completion.
The IFU is evaluated in formative studies to establish that the formats and content is easily read and understood prior to summative HF validation testing. If the formative evaluation has been carried out successfully, the subsequent validation testing should result in good performance and few-to-no use errors with the finished, to-be-marketed product. The realism and completeness of validation testing should support claims of safe and effective use by the people who are representative of the intended users under expected use conditions for essential and critical tasks.
The Center for Drug Evaluation and Research (CDER) has asked to consider including a task to simulate complete device failure in the summative HF study. It was explained that users' reactions to non-standard scenarios (broken device, empty device, piece missing, etc.) are evaluated in design studies during development to enhance the user interface and create user instructions. The rationale was provided that the summative HF study is validation testing and is conducted to establish that the device can be used without patterns of preventable use errors, and it requires the device to operate normally. The FDA accepted the rationale that validation should not be performed at the complete failure extremes, analogous to the accepted rationale that process validation for a drug substance manufacturing process should not be run at the edges of process failure.
Depending on (1) the complexity of the injection system, (2) prior experience with device, and (3) the focus of the HF study plan, a wide number of formative studies are conducted for a given combination product. Multiple (up to six) formative HF studies have been run for a relatively simple injection device prior to running the summative HF study. Alternatively, for a well understood drug/device product where previous experience could be applied, going straight into summative HF study without prior formative HF studies has been acceptable. However, Lilly has been in the unfortunate position of having a completed summative HF study “turn into” a formative HF study (e.g., an additional summative HF study was required). After running two formative HF studies that had not identified new use errors, the summative HF study identified an unforeseen new use error. The new use error observed was then mitigated through device design modification, revisions to the IFU, and changes to the product packaging. A new summative HF study was run and the use error in question was not observed, nor any new use errors identified.
The device testing conditions allow for realistic device–user interactions by use in the final device design labeling and incorporating expected use conditions and environmental effects (e.g., lighting, noise levels, and activity). This includes providing participants with the combination product as they would receive it (e.g., in its original packaging, with all documentation) and allowing participants to use the combination product as they normally would (e.g., without interference from test facilitator). One key learning from HF studies is how the user/moderator interactions can potentially affect study results. One should not underestimate the actions of the study moderators and observers in altering a user's actions and causing artificial results. When Lilly first began running HF studies, there were so many observers watching the participant that the person became very nervous about making a mistake. The participant would not make a move in the fear of what the observers might think or say. Specific to autoinjectors, one participant was so nervous with all those eyes watching, that the pen injector button was not pressed down with any force. The participant thought the button was depressed, but actually did not engage the driving mechanism and the injection was not performed. Only when the pen was raised from the skin did the user depress the button fully, injecting placebo solution into the air.
Training Decay
A topic regarding study execution that would benefit greatly from a standard approach is the appropriate period of time to wait between training on a task and performing the actual task. This is training decay and is meant to be the allowable time that is representative of what happens in real life between training by a health care professional (HCP) and the patient performing the task (e.g., by themselves at home). An expectation has been to allow at least 1 h between training period and running the study. This 1 h delay has been accepted by the FDA for those products taken up to once daily. However, the 1 h training decay was unacceptable when the product administration is greater than one day. FDA stated that a 1 h training delay is not representative of a real-life scenario for a product dosed at a 2 or 4 week interval. Recent feedback on training delay noted that ideally a training decay period should be representative of the actual dosing interval of the product; however, the FDA did accept a minimum decay period of 24 h for a twice monthly administration.
Submitting HF Protocols
The importance of ensuring that the usability study provides a complete assessment of the potential use errors and mitigation plans has led to regulators recommending (and sponsors wholeheartedly agreeing) to submit the summative HF protocol to the FDA for review prior to the sponsor running the study. This has allowed interactions with experts within the Center for Devices and Radiological Health (CDRH) and CDER who have shared pertinent advice based on their experience across a variety of combination products. When the HF summative study involved the injection of patients with placebo, the study protocol may be sent to the Office of Drug Evaluation within CDER under the Investigational New Drug application as a courtesy. The HF study protocols may be submitted as separate Type C meeting requests, as well as in pre-NDA/BLA meetings through pre-submission meeting mechanisms. The FDA/sponsor venues for receiving HF feedback have been face-to-face, teleconference, and written correspondence. In conjunction with submitting the protocols, the product labels and IFUs are included, as well as shipping product samples (3–5 of each combination product including the product carton). In some cases videos demonstrating use of the device have been provided to help the FDA reviewers understand device usability. Sending actual device samples and/or demonstrating the device usability through a video has been “worth a thousand words” in introducing the device to the reviewers.
The FDA has continued to advise sponsors to consult the FDA early in the HF testing process, even prior to formative HF study plans. In submitting the formative HF protocols to the FDA in conjunction with a planned FDA product discussion, sponsors can request feedback on specific early-phase HF combination product topics and not on the entire formative HF protocol. Lilly has submitted and received feedback from the FDA on several formative HF/usability study protocols seeking guidance/comment-specific feedback on critical tasks selection, product differentiation criteria, and appropriate design mitigation strategies to reduce potential use errors.
End User Groups
One of the most challenging aspects of HF study plans has been defining the appropriate distinct user groups that will be using the drug/device product. User diversity continues to increase. People are living longer and surviving more serious injuries and illnesses than ever before. The increasing prevalence of chronic conditions and patients being released from hospitals sooner has led to more health care moving into the home, where the combination product is used by professional caregivers (e.g., physicians, nurses, nurse practitioners, home care aides) and non-professionals (patients providing self-care, family members, friends).
The June 2011 draft HF guidance states that the FDA views populations as distinct when their abilities or the nature of their device interactions are expected to be different (9). For example, some devices will have users in different age categories (pediatric, adolescent, adult, or geriatric); others will have users in different professional categories (e.g., nurse, non-professional family-member caregiver). It is critical to identify the distinct user groups that will be included in the HF studies. FDA feedback has been based on the critical differences in capabilities that would make a distinct user group. HF studies may show no differences in capabilities and supported combining user groups when moving into the summative HF study. For devices with multiple user populations that have different personal characteristics, it is best to try to test the maximum number of participants that budgets and schedules allow.
The June 2011 draft HF guidance stated a suggested sample size of 15 people is sufficient to find a minimum of 90% and an average of 97% of all problems based on the 2003 Faulkner paper on individuals with varying levels of experience testing software (17). If users with distinctly different characteristics (e.g., use responsibilities, age ranges, skill sets, or experience levels) will use the device, validation testing activities should include 15 from each major user group. The most important aspect of sampling may be the extent to which the test participants correspond to the actual end users of the device, which requires that the manufacturer accurately identify and describe its user populations. As an example, CDER provided protocol feedback to double the HCP study size arm by requesting a physician arm be added to the proposed nurse arm. Evidence that physicians, while qualified, almost never performed the injection satisfied the FDA that physicians did not need to be included.
The June 2011 draft HF guidance requires test participants must be American citizens (9). The rationale from the FDA is that they are responsible to U.S. consumers and therefore users tested must be representative of said group. Rather than entertain arguments from manufacturers about why Canadians or English-speaking Europeans are adequate surrogates, the FDA just made it a requirement that HF testing be done in the U.S.
Additional feedback and follow-up discussions with FDA have occurred on the following user groups:
Impairments
The design of the pen injector or auto-injector has taken into account all potential users of the device (self-administered by patients or administered by HCPs or care givers) and the abilities of the distinct user groups. The design is meant to be accommodating to all potential user groups for a given disease state. Therefore, each medical symptom related to the given disease state represents unique user profiles that can affect safe and effective use of the product. The design of a device can be modified to be more accommodating of impairments of the user.
For example, people with diabetes often have some degree of retinopathy (a degenerative disease of the retina), which causes impaired eyesight. These users have difficulty reading displays, such as small numbers and text, or if the contrast is low; therefore magnification or color contrast of that display may be of great benefit. The same can be said for neuropathy, which can cause impaired hand movement. These users have difficulty with hand dexterity, such as dialing a dose or pushing down the dose button. Thus the design takes into account potential impairments to eyesight to make displays easier to read, or impairments to hand movement to make the injector easier to hold/handle.
Although the main differentiation for the product should be the product name on the label, it is recognized that other attributes such as color on the pen body or label color may be used to further differentiate the product. Differentiation is important for products where a patient may be using more than one pen injector, or where multiple pen injector users are within a single household. For HF studies evaluating the ability of participants to choose the correct pen injector where color is used as one of the differentiation attributes, the recruitment of colorblind participants may be necessary. Incorporating colorblind users allows full evaluation of how easily the correct pen injector is chosen from a group of pens, even when the color differences are obscured or negated.
Trained/Untrained
Mixed responses have been received across FDA centers on the need to include both trained and untrained groups as unique user groups in HF formative and/or summative studies. The feedback from CDRH has been to “say what you are going to do and then test for what you said you would do”. CDRH has advised that if a sponsor plans to incorporate formal training in the commercial setting, then the HF studies needs to only validate the training arm scenario. If no formal training was planned for commercial use, then the summative HF testing would only have an untrained scenario. The June 2011 draft HF guidance does not specify experience as a criterion for a distinct user group nor suggest that a summative study include different training levels, only that training be consistent with that expected in commercial use. Formative studies and risk analysis are used to determine if mitigations such as a formal training program are necessary to address unacceptable use errors.
CDER, however, takes a different approach in the testing of participants. CDER has less confidence that training will actually occur in real life, even if formal training is planned. CDER expects both a trained and untrained arm be incorporated as unique user groups in HF testing. Figure 2 shows a breakdown from a graph provided by the FDA in response to feedback for a submitted summative HF protocol. CDER recommended that each user group in the validation study include a minimum of 15 trained and 15 untrained users, which resulted in a minimum of 30 participants per user group, thus doubling the size of the study. Validation studies for injection devices will typically include at least four to six distinct user groups and each one will include at least 15 participants. Thus, typical injection device HF validation studies will include a minimum of 60–90 participants.

FDA response for the user group to be tested for trained/untrained.
Naïve/Experienced
Based on FDA feedback on several summative HF protocols, FDA has recommended the validation studies include specific numbers of injection-experienced vs injection-naïve participants. CDER has recommended the validation study include a minimum of 15 injection-experienced and 15 injection-naïve participants per distinct user group. Injection-experienced and injection-naïve participants should only be required in summative studies if these groups were identified in formative HF studies to have use characteristics that are distinct from other user groups. This has led to identification of several potential user groups based on their level of experience with injections, devices, and even type of device. An example of user groups evaluated in formative HF studies for insulin pen injectors were (1) injection-naïve, (2) injection-experienced, (3) insulin injection–experienced, (4) device injection–experienced with vial/syringe, and (5) device injection–experienced with pen. Table III provides an example of the cohort groups included in a formative HF study evaluating users' ability to understand and execute a particularly difficult volume conversion concept that was described in the IFU. The results provided insight into how best to describe the concept through text and graphics and number of use steps in the pen injector IFU.
Example of Cohort Groups Studies for a Formative HF Study
Despite test coordinators' best efforts, participants can be unrealistically well trained, capable, or extremely careful. When people are observed they often try to do their best; they tend to follow instructions more carefully than when they operate the device independently. The opposite is also true with respect to attention to detail. Participants can be overconfident in their abilities to use the device. One group of experienced users that has generated an unusually high number of use errors are HCPs. HCPs can be quite confident in their abilities and so sure they know how to use the device, often from previous experience with a perceived similar device, that they discard the instructions and jump right into performing the use steps. A high number of use errors can result from this overconfidence.
A remaining concern in running HF studies for injectables is not to bias the testing results (i.e., to find lower use errors than is realistic) due to experience gained by naïve user groups during the study. The FDA has provided feedback on multiple HF protocols recommending revisions regarding the order in running the tasks, or using a whole new set of participants to avoid “training” the user during the study. The FDA has stressed the need for keeping the user “fresh” as they move through critical tasks, and/or repeating tasks in the use of multi-dose injectors. This has become a complex endeavor, but it is possible through careful planning to minimize prior learning of participants for performing subsequent tasks.
Usability Testing and Clinical Experience
Conflicting guidance has been received from FDA centers on the expectation of usability study requirements in support of acceptable testing for the drug/device combination product. In general, the medical experts within the Drug Evaluation divisions in CDER tend to address a device performance issue by requesting clinical trial data, while the engineering/mechanical experts within CDRH tend to address the device performance issue by requesting HF usability studies. A clinician naturally takes a clinical approach for the assessment and an engineer will naturally take a mechanical/ergonomics approach to gathering data.
The Drug Evaluation divisions within CDER have repeatedly inquired about collecting usability information during clinical trials. CDER has requested “real-life actual use data to support marketing approval” and “usability of your product presentations during the clinical trial” but has not provided examples of how this should be executed nor the outcomes the FDA will require. The FDA has made this request as an additional study to conducting the HF validation study and without having reviewed the planned simulated use summative HF study plan. This type of request was included in the May 2013 draft guidance led by CDER's Division of Pulmonary, Allergy, and Rheumatology Products (15) that made the following recommended study requirements for a device change: “The extent of clinical data needed to support such changes depends on the nature of the change and the development stage. For example, a transition from a prefilled syringe to an autoinjector delivery system involves the following, at a minimum: (1) human factor studies to evaluate potential use-related risks of the modified combination product; (2) a pharmacokinetic bridging study that demonstrates similar delivery of the drug product to the same biospace across a range of body weights; and (3) real-life patient handling experience to assess device performance as discussed above” (emphasis added).
However, there are limitations to conducting a usability assessment in a controlled clinical setting. Clinical trials for injection devices are seldom conducted in a real-world settings such that it may be difficult to glean normal intended device use conclusions from the clinical outcomes. Adverse events and complaints from clinical trials are evaluated for potential relationship to the injection device; however, patterns of use failure or use difficulties usually cannot be identified by adverse event or complaint reports alone. In addition, devices used in simulation testing are generally new and in good operating condition and it may be difficult to simulate the effects of long-term use. The user in a clinical trial setting would not be expected to use the device long enough to experience problems that arise more infrequently.
To ensure the drug is delivered in the same way as the previous administration (vial/syringe vs autoinjector), the device may be incorporated into a clinical bridging study. The key point of the clinical bridging study, however, is to evaluate clinical outcome of the drug administered by the device, rather than usability, which is the purpose of the HF testing. The HF studies (simulated use) are the most rigorous means to assess usability. CDRH has expressed concern with assessing clinical and simulated use endpoints in the same study because it could overly complicate the study. CDRH recommended that simulated use testing to evaluate usability be separated from clinical trials where trained observers watch the participants completing specific tasks with the device to uncover patterns of use failure and use difficulties.
Simulated vs Actual Use
CDER has recommended performing validation testing under conditions of actual clinical use be considered when simulated use validation methods appear to be inadequate. CDRH's June 2011 draft HF guidance states, “Due to the nature of some types of device use or use environments that may be particularly challenging or poorly understood, it might be necessary to validate a device under conditions of actual use in a clinical study.” Actual use should only be required when the device use or use environment are poorly understood. However, injection devices generally don't have particularly challenging or poorly understood use or use environments. Simulated use studies (e.g., injecting into a pad placed against the injection site rather than injection into the body) are typically acceptable to appropriately identify use errors and assess user interactions with the device. Simulated use studies have been acceptable to the FDA because injection surrogates are appropriate in representing injection sites and patient handling, and the results of the testing can be considered realistic of actual use.
Placebo Injections
As stated above, injection devices typically should not require placebo injection during the HF validation study, as simulated use with an injection pad has been sufficient to ascertain usability assessments of the drug/device combination product. However, the FDA drug review division has asked for justification of why the use of injection pads would be appropriate for a summative autoinjector study rather than a placebo injection. Lilly was able to successfully argue that the injection pad was fully representative of patient use and acceptable for simulated use. Placebo injections were not required.
An exception to the use of injection pads was in running the first summative HF study with a new autoinjector platform. CDER recommended the summative HF study involve placebo injection and not the use of injection pads due to the lack of manufacturer's experience with the device. In this case, the summative HF study was performed with placebo injections. Other concerns against using injection pads have been that the pad might impede product use or the participants do not perform the task as intended because they deemed the simulated injection to not be realistic. In one case a participant was asked about a use error where he/she did not keep the injector on the injection pad through completion of the dose (i.e., early lift-off). When asked about the use error, the participant said, “Well, I would never have done that if it had been an actual injection”. It is therefore important to ensure that a simulated use is truly representative of what will happen when the product is used in the real world, and placebo injections may be useful to eliminate that concern.
Submission of HF in Marketing Application
The relevant HF study information of testing results, risk management, and design and instruction optimization is submitted in the marketing applications (e.g., U.S. NDA/BLA and European Union Dossier) for injectable combination products. The HF information demonstrates that the sponsor has adequately addressed the needs of the intended user groups and optimized the device design to assure the drug/device combination product is safe and effective. An outline of the sections of the HFE/UE report to be submitted to the FDA is shown in Table IV and follows exactly the FDA's June 2011 draft HF guidance expectations (9). The report size varies, but as a guide to the level of detail needed to highlight the major HF considerations, issues, resolutions, and conclusions, the reports have been 50–70 pages. The HFE/UE report can be added as an attachment to the submission, or the seven sections of the report can be lifted and placed in the corresponding 3.2.R Regional section of the marketing application following the eCTD format in both the U.S. and European Union.
Sections of the Human Factors Engineering Report
As pen injectors, pre-filled syringes, and autoinjectors have become platform delivery systems for multiple products, Lilly has reached agreement with the FDA to implement Master Files for Devices (referred to as a MAF by the FDA) for these device platforms. An MAF has largely been used to submit information to the FDA by the MAF holder of its own confidential data without it being disclosed to anyone outside the FDA. However, the MAF may also be utilized when several different product applications use a common platform (18). The MAF will include the device information for device platforms referenced across multiple product NDA/BLAs for the delivery system. The NDA/BLA contains all information on the drug-related information, including all the container/closure test results, and the MAF contains all information specific to the device: assembly, materials of manufacture, HF reports, and so on. Information pertaining to both drug and device would be cross-referenced in the NDA/BLA and MAF. Post-approval changes that require regulatory reporting would be submitted to the NDA/BLA, but those changes below the level of supplement reporting would be revised in real time in the respective device MAF without the need to report them in multiple product NDA/BLAs.
Instructions for Use
The design of device labels and instructions begins early in the device development process (19). The IFU is first written as the device concept evolves as a result of device prototype testing and HF evaluation. The final validation as to the acceptability and adequacy of the labeling for device labels and instructions occurs during the summative HF validation study, which is completed before submission. Instructions, labeling, and training can influence users to use devices safely and effectively and are critical HFE/UE considerations for safe device use. Because these “fixes” rely on the user to remember or refer back to the information, the approaches are less effective than modifications to the design of the user interface. Still, mitigation of use-related hazards through well written and illustrated IFU can be quite effective and valuable.
It is essential to assess user performance based on device design, but it is also important to determine whether users can acquire the necessary knowledge from the labeling materials. The importance of getting the instruction optimized is recognized by both regulators and sponsors. For injectable products, the instructions may go beyond just ensuring clear statements and graphics, but may need to convey concepts like concentration conversions and volume/unit additions. This has required a more focused attention on IFU content, requiring an increased knowledge of educational instruction to optimize content and the need to conduct additional formative HF studies to evaluate that prescriptive content is effective in the reduction of dosing errors.
An area that would benefit from a common FDA centers approach is the acceptability of content and validation studies used in support of the IFU. Feedback on IFUs has been inconsistent across CDER and CDRH. An example was an FDA request late in the approval process for modifications to labeling that had already been successfully validated through the summative HF study. CDER's Division of Medical Policy Programs required the IFU for an injection device to have significant text revisions to several instruction steps, and required the addition of new figures and revisions to existing figures.
This example points to the need for FDA review of IFUs to occur early in the review cycle so that IFU wording and figures can be finalized with complete review of justifications and consideration of alternative options. Obtaining specific IFU change requests from the FDA during review negates the HF validation results. Change requests should only be required if the FDA has published information that the wording as submitted would cause patient harm and has data establishing that FDA-recommended changes have been validated. The FDA needs to allow manufacturers to utilize labeling that meets the regulatory requirements and whose content has been appropriately validated in HF studies. There is legal precedence that the sponsor owns the label and the sponsor, not the FDA, is libel if there are adverse safety events due to the label (20).
The HF assessments have also included the optimum presentation to impact usability of the IFU document—single sheet leaflet, accordion style fold-out, or bound booklet—and even the way a sheet is folded and placed into the carton (to best catch the eye of the user). The HF study results have shown that some participants have completely discarded the IFU or may have only partially opened the IFU before beginning the use steps. Participants have made suggestions on how to fold the IFU so that users could immediately see the figures for the use steps as soon as they pull it out. In an HF study where the product would be administered by a parent, one father who had discarded the IFU recommended during the interview process that immediate sight of the graphics would grab parents' attention and make it less likely that parents would ignore the IFU.
The rise of instructional videos placed on the web is a growing consideration in context to HF studies and the IFU. In this day of YouTube videos, the prevalence of “How To …” videos has blossomed. Quick reference codes printed on package and IFUs has allowed smart phone users to go straight to the product/company web site and download instructional videos and promotional materials. There is also a growing trend of individual patients producing their own instructional videos and uploading them onto YouTube. These instructional videos on the web, which have been produced by individuals not affiliated with the manufacturer, appear to do a good job of instructing the user. This is presumably due to the information in the video being based on the manufacturer's IFU (and thus the HF study results).
Promotional Claims
Once the commercial product is approved and ready to reach the marketplace, there is the strong desire to be able to make promotional claims on usability such as “Easy to Use” or “Simple to Use”. The question has been asked by marketing groups of what HF testing is needed or could be used to help support the sponsor in make promotional claims. Differentiation between injectable products based on ease-of-use claims is a growing area of concern with the hopes of being used as a competitive marketing advantage. Injectable combination product web sites often carry claims on usability—easy to grip, easy to read display, 3 easy steps—with a reference to the claim of “data on file”. It is unclear what the standards are for data requirements and what should be the acceptance criteria to make such usability claims. Questionnaires are often given to assess ease of use or ease of training with sponsors soliciting participant feedback on the design of device, labeling, and training. Forced-choice questionnaires or Likert rating scales are used to collect limited ease-of-use information. But what acceptance criteria score is needed to support the ease-of-use claims is not well understood or standardized. The FDA has stated its concern that such questionnaires can bias responses to promotional claims. The FDA has stated generating evidence to support promotional claims like “easy to use” and ”easy to learn” are unlikely to be allowed in the labeling on the basis that these claims are subjective and vague.
Post-Approval Changes
The focus on continuous device improvements and incorporating design iterations is much more prevalent with an injector device than making corresponding drug changes through formulation and/or manufacturing processes. It's an expectation from both regulator and sponsor that continuous design improvements will continue throughout the life cycle of the device to improve usability and performance. Injection devices are iterated upon for various reasons including consumer preferences, manufacturing productivity improvements, device feature improvements, and mitigation of customer complaints. It's common for new injection devices to be based upon a previous device “platform” but incorporate device modifications to adapt it to a new dose or intended user group(s).
The decision trees contained within the FDA's 1997 guidance for devices (21) cleared under the Food, Drug, and Cosmetic Act 510(k) regulations provide the relevant guidance on the reporting of post-approval device changes for injectable combination products. The decision trees have been very helpful in understanding the types of device changes FDA has determined to have the potential to adversely affect the drug/device with respect to patient safety and effectiveness. The decision trees have been especially useful in evaluating revisions to material of use and component parts. The potential adverse impact can be put into context of affecting the “drug flow path”—either through contact with the drug solution (e.g., staked needle, plunger) or flow (e.g., injection rate, completion of dose). However, as injector combination products have the drug as the primary mode of action and thus CDER as the lead division, the expectation is that post-approval guidance and reporting for drugs through submission supplements and annual reports be considered. It still holds that the drug flow path is the main criteria in the evaluation.
With respect to HF testing in support of post-approval changes, CDER expects risk assessments to determine the need for subsequent HF studies. The FDA expects risk assessments to be conducted for any device modifications to determine if new functional and/or simulated use risks are identified and to determine if new or different use errors could occur. If new use errors are likely to occur based on the risk assessment, then those aspects should be evaluated via HF studies. Aspects of the previous device version or platform that were not changed should not require further HF evaluation. This is consistent with CDER recommendations that subsequent bench testing and/or HF validation studies might be needed. However, the FDA has not yet provided a decision framework for evaluating changes to the device, the intended users, or use environments.
Conclusions
HF usability testing is critical in the assessment and subsequent mitigation of use errors for injectable combination products. The earlier the HF work is incorporated into product design the better, as identification of the intended uses, users, and use environments is crucial in the process of design development and evaluation. The use of formative HF studies to evaluate and mitigate use errors and define distinct user groups has paid dividends in the subsequent validation of usability through the summative HF studies. A well designed HF validation study has critical and essential tasks prioritized with potential use errors evaluated and mitigated through design modification and clear IFU. The injectable combination product HF information is included in the submission and provides clear evaluation of the use errors and rationale for any remaining residual risk being outweighed by the benefits of the delivery system. The HF studies have provided the needed assurance of appropriate product usability without the need for tacking on a clinical patient device handling experience that does not have the observational acceptance criteria to adequately measure device usability. HF studies not only demonstrate the positive benefits vs risk of the device, but ensure the development and commercialization of safer, more effective and usable drug/device injectable combination products.
Conflict of Interest Declaration
The author is an employee of Eli Lilly and Company and declares no other competing interests related to the article.
Acknowledgements
The author would like to thank April Berry, LeeAnn Chambers, Debra Conner, Patricia Cowall-Hanover, James Kershner, Amy Kovacik, and Mark Marley for their insight and thoughtful discussions on the planning, execution, and reporting of HF studies and for their review and comments on this paper.
- © PDA, Inc. 2014




{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- Renewed Emphasis on HF
- Scope of HF
- Risk Evaluation and Mitigation
- Formative Evaluations and Summative Validation HF Testing
- Training Decay
- Submitting HF Protocols
- End User Groups
- Usability Testing and Clinical Experience
- Submission of HF in Marketing Application
- Promotional Claims
- Post-Approval Changes
- Conclusions
- Conflict of Interest Declaration
- Acknowledgements
- References
- Figures & Data
- References
- Info & Metrics