
Abstract
The inability to detect endotoxin using compendia methods is a potential safety concern for patients due to the lack of endotoxin removal capabilities at the fill–finish stage in typical aseptic biologic drug product manufacturing. We have successfully demonstrated endotoxin challenge study recovery methodology using mammalian cell–produced biologic drug products and drug substances in citrate, histidine, phosphate, and sodium acetate buffer formulations containing polysorbate, challenged with an endotoxin analyte, for up to 6 months of storage. Successful recovery was similarly demonstrated for a preserved, peptide-containing drug product formulation. To isolate a potential masking—or low-endotoxin recovery—source, additional studies were performed to evaluate factors including product manufacturing contact surfaces, drug product matrix with and without polysorbate, individual matrix components, protein concentration, reagent suppliers, an orthogonal test method, and storage conditions. In all cases, acceptable recoveries were observed. Bacterial endotoxin is known to be chemically stable at physiological conditions. Purified endotoxin in aqueous conditions is likely to self-aggregate or bind to surfaces. Neither the nature of, nor the storage conditions of, the studied formulation matrices were shown experimentally to render the challenge endotoxin biologically inactive. The results highlight the importance of appropriate study design in assessing the recovery of endotoxins.
LAY ABSTRACT: Bacterial endotoxin is a Gram-negative bacterial cell wall component that is harmful to humans at threshold concentrations, and it is not expected to be in aseptically-produced pharmaceutical medicines. It has been suggested that endotoxin cannot be detected over time in certain biopharmaceutical drug product formulations containing citrate, phosphate, and polysorbate components via an unknown masking mechanism. We have generated and present data here that indicate that endotoxin can be recovered in a variety of matrices, and under various experimental conditions.
Introduction
The control of pyrogens, but especially bacterial endotoxins, in the production of parenteral pharmaceutical products is one of many requirements to assure patient safety. Bioproducts, especially those produced in mammalian cell culture, have numerous manufacturing steps involving complex growth media and buffer matrices that promote microbial growth. Therefore a comprehensive microbial and pyrogen control/monitoring strategy is required to assure consistent removal and control during the purification steps for the drug substance and formulated drug product. The primary pyrogen source is endotoxin from Gram-negative bacteria potentially present in the microbial flora of the manufacturing equipment and aqueous matrices. The purification processes for drug substances are designed to minimize adventitious microbial contamination, but they are not sterile. Therefore bioburden and endotoxin monitoring of the in-process stages of purification to the final drug substance, as well as the formulation of product at the aseptic fill–finish steps, are routinely performed with results evaluated against established limits. The techniques described in United States Pharmacopoeia (USP) General Chapter <85> (1), and harmonized with the European (2) and Japanese Pharmacopoeias (3), are typically employed for endotoxin monitoring of these various matrices. Part of the compendia method verification suitability for each matrix type requires demonstration that the sample, as diluted and prepared for testing, is within biological pH range, and that a positive control derived from a control standard endotoxin (CSE) can be recovered within 50–200% when applied to the test dilution. The CSE is a chemically purified lipopolysaccharide (LPS) preparation derived from an Escherichia coli strain used to produce USP reference standard endotoxin (RSE).
Endotoxin challenge studies are designed such that samples are challenged with the analyte in the final sample container and tested initially and over various time intervals to evaluate endotoxin recovery. Other experimental parameters, such as endotoxin source, challenge concentrations, and recovery criteria, are defined in the experimental design. The 2012 U.S. Food and Drug Administration guidance (4) recommends that protocols designed to demonstrate stability of assayable endotoxins contents should consider the source of endotoxin used in these studies, as native endotoxins might react differently than CSE. The guidance publication pre-dated the low endotoxin recovery (LER) issue, however, the requirement to perform endotoxin challenge studies have been interpreted from the guidance.
The following experiments were designed and executed to understand the factors that may lead to recovery issues when challenging endotoxin directly to a sample solution, and then analyzing the sample over time using the compendia-verified test method. Public presentations (5) have indicated that the LER phenomenon is of specific concern for biologics in citrate and phosphate matrices containing polysorbate.
The compendia photometric kinetic chromogenic assay was used to evaluate various drug products and components when challenged with a laboratory-prepared naturally occurring endotoxin (NOE) or LPS. We have demonstrated recovery in citrate and phosphate matrices containing polysorbate in addition to other biologic drug product formulation matrices. The use of a NOE as a model challenge analyte is superior to LPS both in performance and as a more representative analyte.
Materials and Methods
NOE Stock Endotoxin Preparation
To better simulate the natural analyte, an E.coli O55:B5 stock from American Type Culture Collection (ATCC) 12014 was grown overnight in several trypticase soy broth tubes. Individual tube contents were pooled into a polypropylene tube and then centrifuged at 3200 relative centrifugal force for 10 min. The broth supernatant was decanted and the cell pellet resuspended in Limulus amebocyte lysate (LAL) reagent-grade water (LRW). The suspension was boiled in a water bath at approximately 100 °C for 10 min. The contents were transferred to a polystyrene tube. Organism kill was verified by analysis of a suspension aliquot on a trypticase soy agar (TSA) pour plate. The TSA plate was incubated at 30–35 °C for not more than 5 days with no growth observed. The stock suspension was assayed for endotoxin concentration by analyzing stock dilutions using the kinetic chromogenic method, and then stored at 2–8 °C in the polystyrene tube. Bowers and Tran (6) describe a NOE preparation using filtration to remove the challenge organism for use in similar studies.
Endotoxin Challenge Study Sample Preparation
The stock endotoxin concentration was quantified prior to use in a challenge study by dilution of the stock onto 0.05 to 5.0 EU/mL or 0.01 to 1.0 EU/mL standard curves using CSE calibrated against the RSE. Based on the observed value, the volume necessary to challenge 0.05 to 0.1 mL of diluted stock to the study matrix was calculated, such that after dilution to the non-interfering concentration in LRW as determined by the compendia inhibition/enhancement test, the challenge concentration would be approximately the mid-point of the chosen standard curve. The mid-point of the curve was chosen due to the increased variability created by the logarithmic reduction of the assay at the high curve concentration, and because kinetic testing is not typically designed to be an endpoint limit test. At each time point, the container was removed from storage, vigorously vortex-mixed at least 5 min, sampled, diluted to the test concentration, and analyzed by the compendia method. We challenged single containers that were amenable to opening multiple times and containing adequate volume (i.e., drug product vials and drug substance sample tubes), whereas replicates were challenged for small-volume items such as semi-finished syringes. The containers were challenged by removing the stopper, plunger or lid; adding the challenge aliquot with a pipette; and re-closing the container. All samples were stored at 2–8°C as is representative of the respective drug product and drug substance storage conditions.
LAL Detection Methodology
The kinetic chromogenic method described in the compendia was used in combination with a Biotek Elx808 absorbance reader measuring optical density (OD) change at 405 nm and a 0.2 OD cutoff. Lonza WinKQCL™ software was used for sample analysis. Lonza LAL and CSE reagents were primarily used, but Charles River Endosafe and Associates of Cape Cod kinetic chromogenic reagents were also evaluated to investigate a potential supplier effect on LER.
The Lonza Pyrogene® recombinant Factor C (rFC) system was used as an orthogonal non-LAL detection methodology. The rFC method is a 60 min fluorogenic end-point test using a Biotek FLx800 instrument with 380/440 nm excitation and emission wavelengths. Data was analyzed using Lonza WinKQCL™ software. A three-part enzyme master mix reagent is used in combination with the same CSE used in the LAL methods, and unknown values are plotted against a positively-correlated curve.
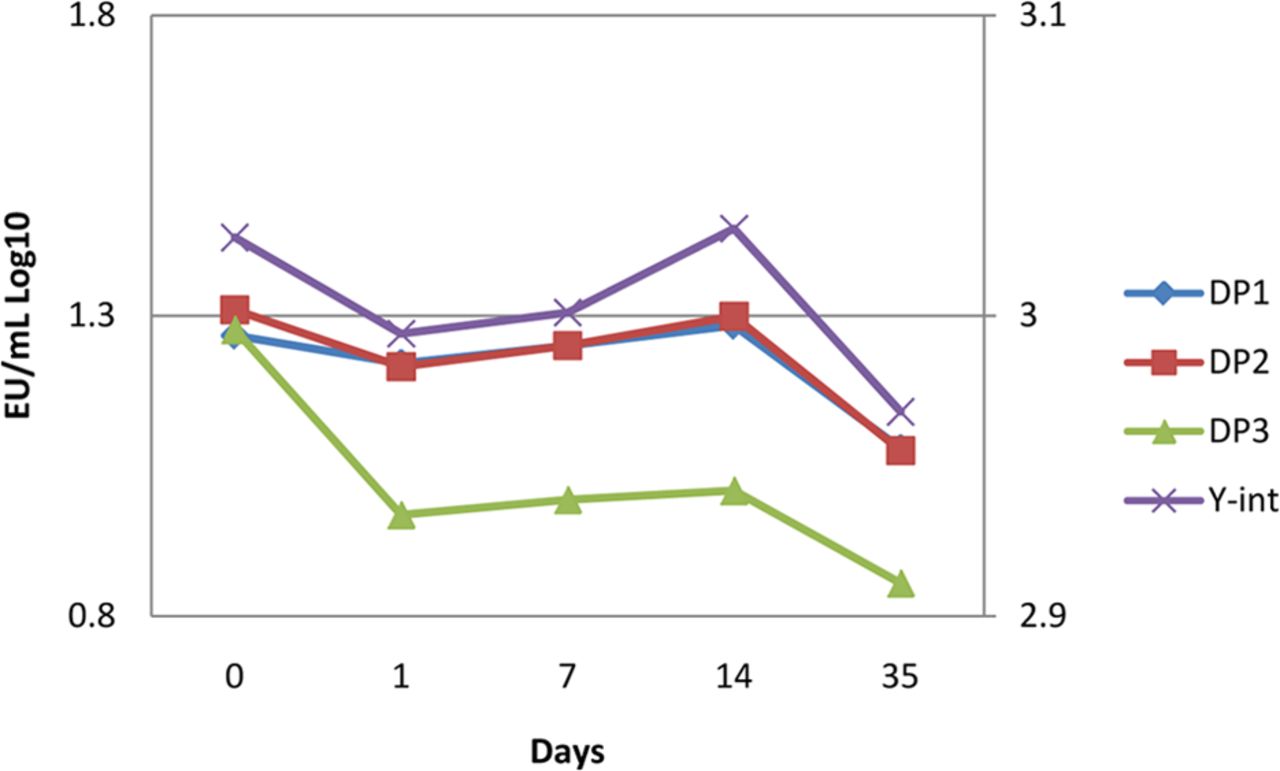
The acceptance criteria for linearity as described in the compendia were used to determine assay and sample validity, in addition to in-house y-intercept, slope, and replicate percent coefficient of variance (%CV) system and data criteria. Recovery acceptance criteria was chosen to be ±0.5 log from the initial time point recovery. This criterion was chosen for (a) alignment with compendia microbial challenge testing, and viral challenge testing; others have used the compendia-derived 50-200% positive control recovery criteria (5, 7); (b) a theoretical demonstration that a 1% y-intercept shift can cause a 36% recovery shift (8); (c) observed recovery shifts due to corresponding standard curve y-intercept shift (Figure 1); and (d) the picogram-level detection capability of the assay.

Y-intercept variability is inherent to the biological nature of the assay. Endotoxin challenge recoveries were observed in three different batches of the same drug product (DP1, DP2, DP3) to shift with the inter-assay y-intercept values. The average y-intercept variability was 0.75% across this data set and corresponded to an average 75% recovery from the initial challenge.
Results and Discussion
Citrate Drug Substance and Product Matrix Containing Polysorbate 80
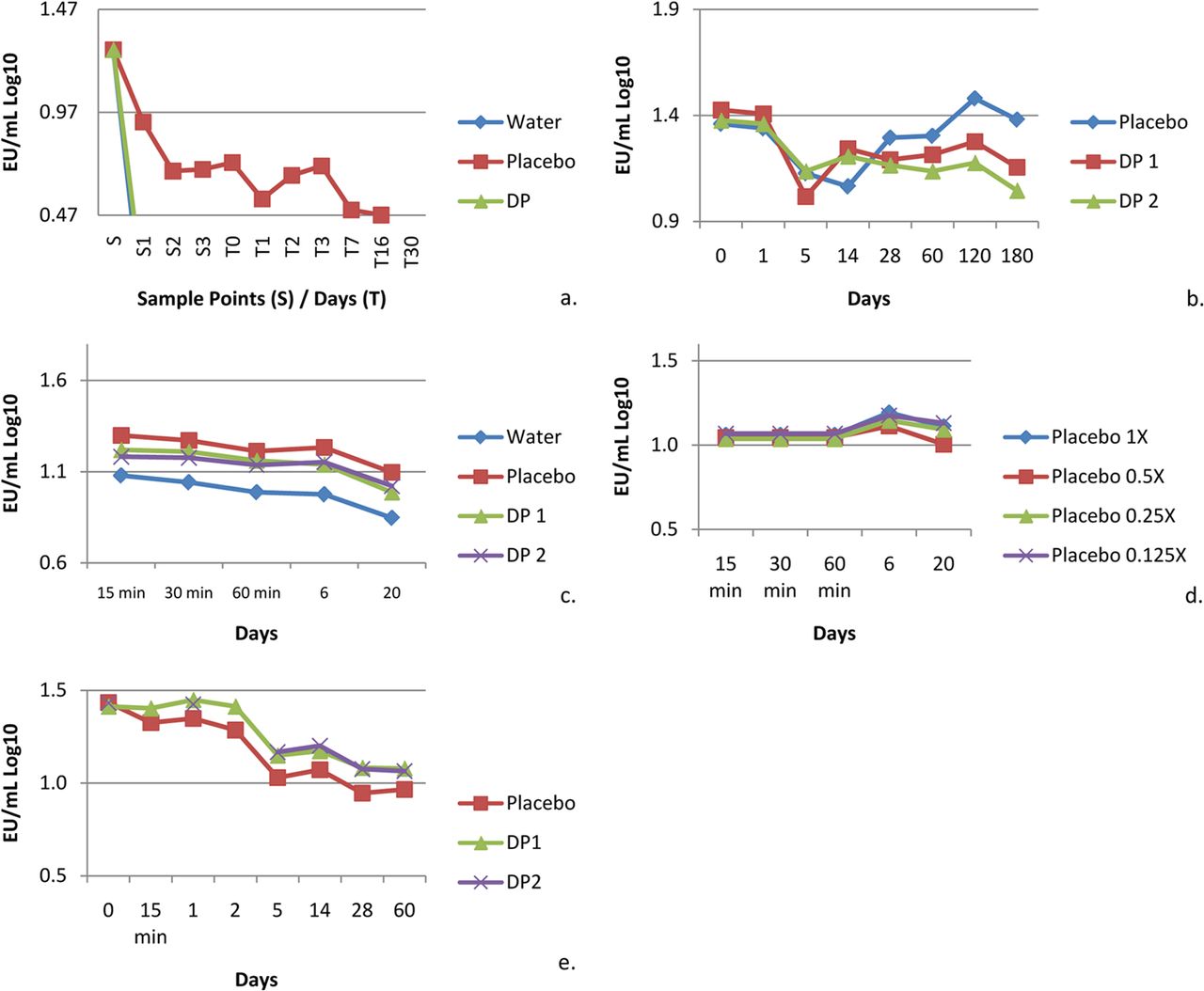
An initial study was conducted to address contact surface loss concerns in which drug product manufacturing was simulated from formulation compounding to pre-filled syringe filling. The target concentration was 20 EU/mL in the drug product, or 0.5 EU/mL after dilution. This drug product contained relatively low protein concentration (<5 mg/mL), 10 mM citrate and 0.02% polysorbate 80. Drug product, placebo, which is drug product formulation without drug substance, and water were challenged with a NOE. Product contact surfaces included stainless steel, polyvinylidene difluoride (PVDF) filtration, polystyrene, silicone tubing, glass, and bromobutyl rubber.
The data from this study indicated that mixing is a critical recovery factor. Endotoxin was not recovered, or was recovered very poorly, during a gentle formulation mix with a stir bar in the stainless steel container. Additional experiments were conducted to isolate the potential source of the low recovery. Similar protein solutions were challenged with endotoxin and held in stainless steel containers for 20 days, in the final drug product container closure system for 6 months, and in polystyrene sample tubes for 60 days. Figures 2a–e summarize the results of these studies. In contrast to the initial experiment, the endotoxin challenge was successfully recovered in all cases. This result reinforces that rigorous mixing is essential to ensure the recovery of endotoxins when measured using the compendia test method.

(a) The initial formulation experiment resulted in poor recoveries. However, recovery was demonstrated in subsequent studies, which indicates no recovery effect due to (b) the drug product and container closure, (c) the formulation tanks, (d) the buffer, or (e) the drug product and polystyrene sample tubes. Two different batches of the same drug product (DP) were used in the study (DP1, DP2).Placebo 1X in panel (d) is the nominal concentration, which was subsequently diluted 2fold to a 1/8 concentration.
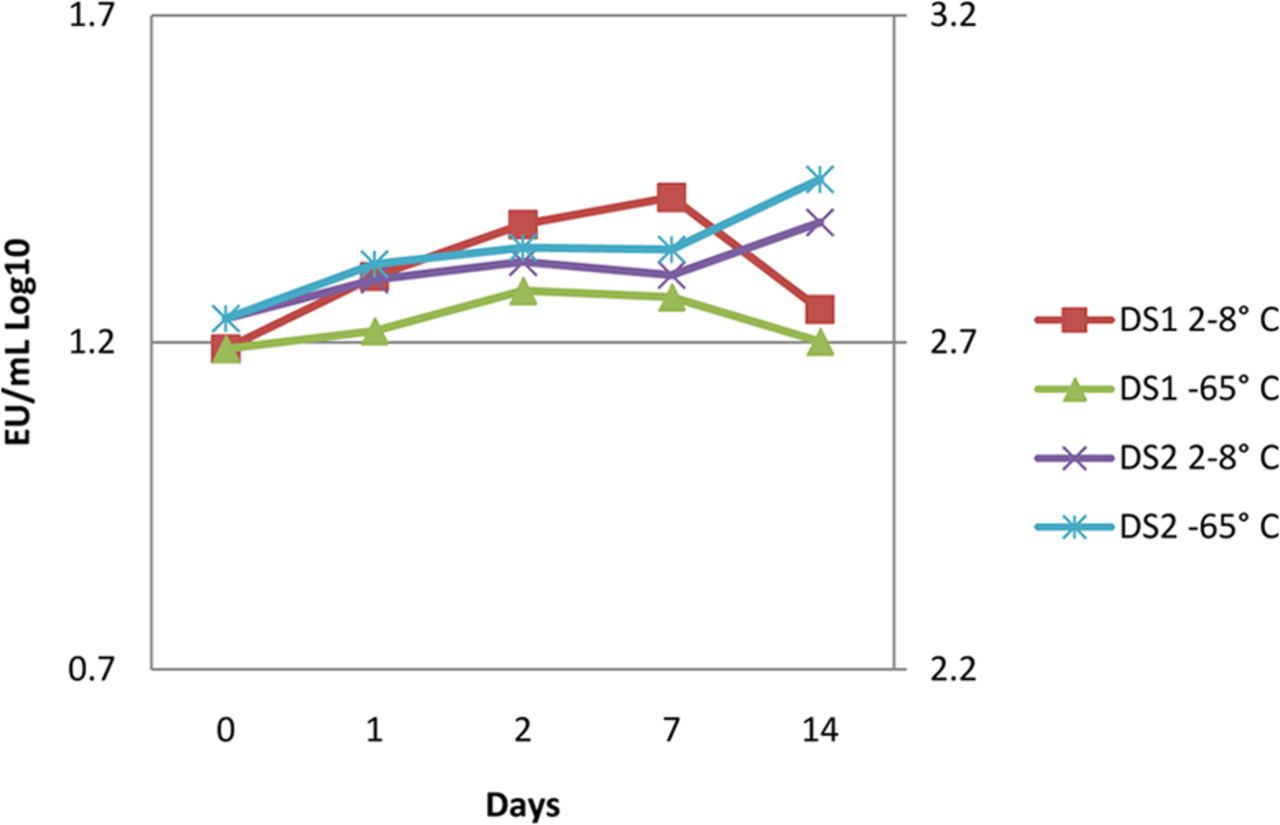
This same study design was executed in the drug substance manufacturing site laboratory. The drug substance was challenged in the final polystyrene sample container then stored at refrigerated (2–8°C) and ultra-low temperatures (–65 °C) for 14 days. The drug substance is in a similar citrate and polysorbate 80 matrix as the drug product but contained approximately 40 mg/mL protein concentration. The results of this experiment are shown in Figure 3 and indicate there is no effect on recovery due to storage temperature or protein concentration.

Endotoxin challenges were recovered from both high (>140 mg/mL, DS1) and moderate (∼40 mg/mL, DS2) protein concentration drug substances in citrate and polysorbate 80 when stored at both refrigerated and ultra-low temperatures, indicating no recovery effects due to temperature or high protein concentrations.
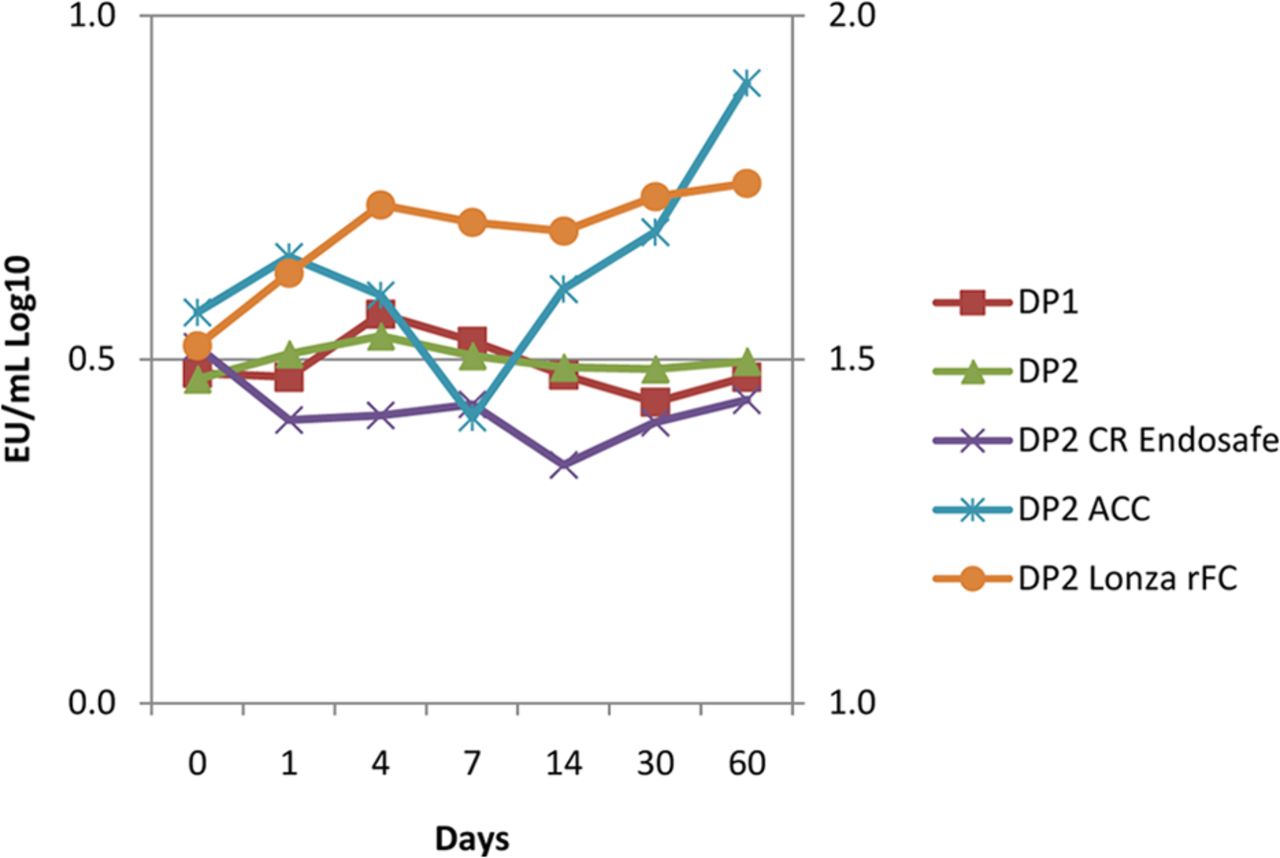
To evaluate the effect of higher protein concentration, additional studies were conducted on formulation containing 80–140 mg/mL protein in 20 mM citrate and 0.02–0.03% polysorbate 80 matrices. Another arm of this study evaluated Charles River Endosafe and Associate of Cape Cod kinetic chromogenic, and Lonza Pyrogene® rFC reagents. The endotoxin challenge was successfully recovered over 60 days using each supplier's LAL (Figure 4). The results indicate that higher protein concentrations do not affect recovery and that there is no apparent LAL vendor-specific effect. Additionally, the capability of the Lonza Pyrogene® system as an orthogonal method to detect the endotoxin challenge was demonstrated.

Endotoxin challenges were recovered from two high-protein concentration drug product (∼120 mg/mL) batches (DP1 and DP2) containing citrate and polysorbate 80 in the container closure system. Recovery was demonstrated in DP2 using conventional LAL kinetic chromogenic methodology across three LAL suppliers, and using recombinant Factor C by fluorogenic endpoint methodology.
Histidine/Glycine Drug Substance and Product Matrix Containing Polysorbate 80
A study was performed at a drug substance manufacturing site laboratory; it simulated the penultimate drug substance manufacturing step through to the drug product vial fill, using maximum production hold times. The drug product and drug substance are in similar matrices and contain approximate concentrations of 10–15 mg/mL protein concentration, 140 mM histidine/glycine buffer, and 0.01% polysorbate 80.
The drug substance was challenged within a stainless steel container, held refrigerated for 24 h, then 0.22 μm filtered into a polyethylene bag and stored refrigerated for a total of 14 days. The drug product was challenged while in a polyethylene bag, held 48 h refrigerated, then 0.22 μm filtered, filled into vials, and stored refrigerated for a total of 14 days. Both the drug product and drug substance were challenged with endotoxin at 20 EU/mL, or 2.5 EU/mL after product dilution in LRW (Figure 5). The endotoxin challenge was recovered in both the drug product and drug substance for up to 14 days of storage.

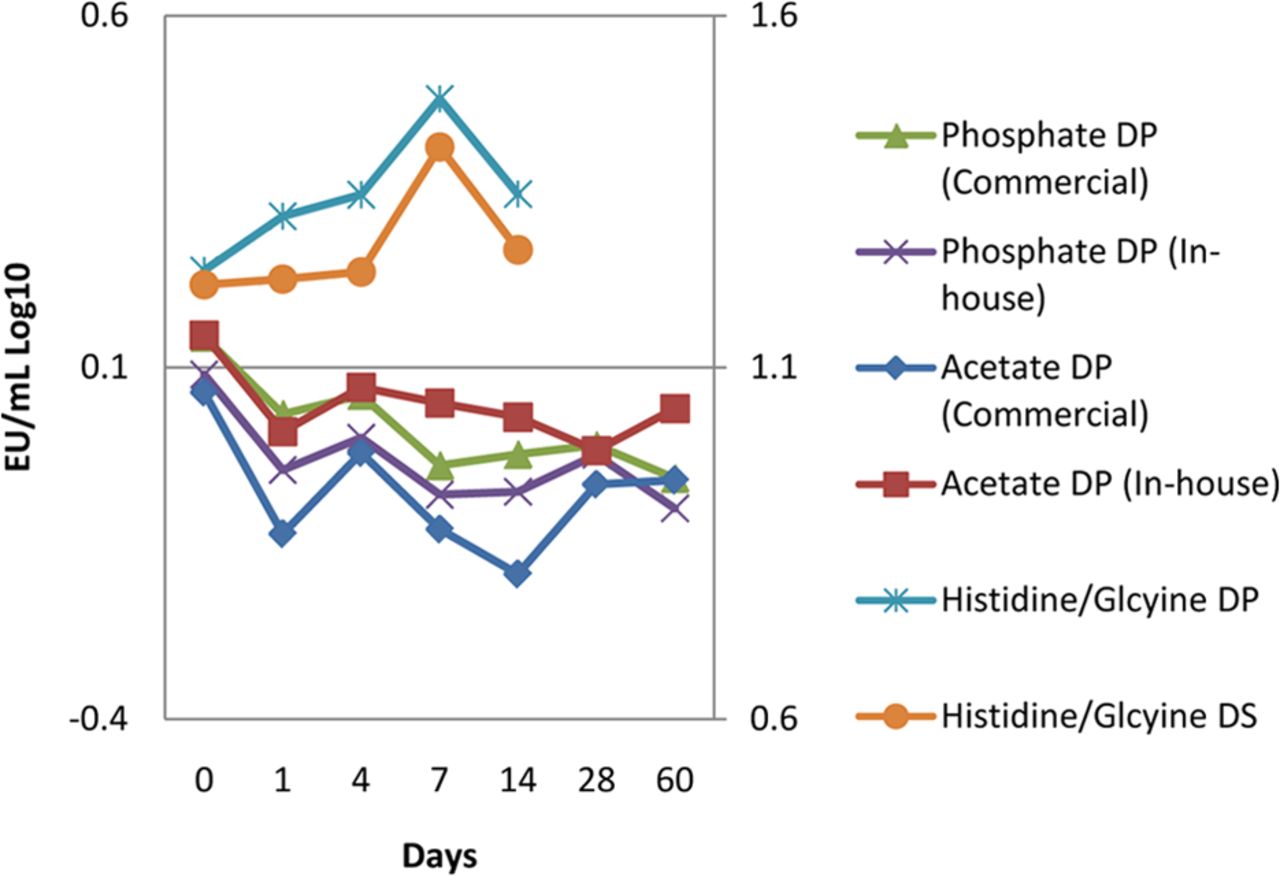
Endotoxin challenges were recovered from additional drug product (DP) matrices including phosphate, sodium acetate and histidine/glycine matrices and a histidine/glycine drug substance (DS) matrix. Both the NOE and LPS were used in the acetate and phosphate studies, and both were recovered.
Additional Studies
Additional matrices were challenged in the final drug product container and held refrigerated. These matrices included antibody drug products in a polysorbate matrix buffered with either sodium phosphate or sodium acetate, and peptides in metacresol-preserved glycerin at low pH, or tris buffer at neutral pH matrices.
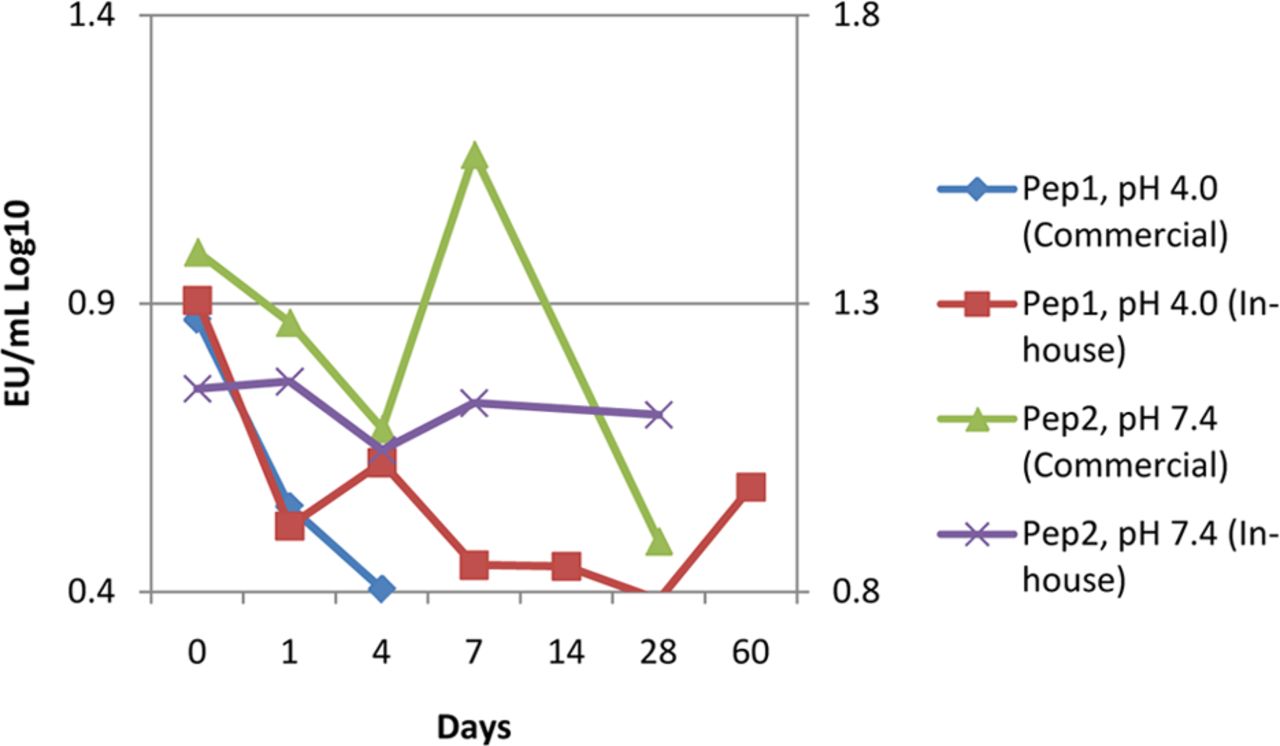
An arm of these studies also evaluated the NOE stock challenge suspension against a commercially available LPS. No practical difference was observed between the endotoxin sources in the phosphate and acetate buffer matrices. Valid, but low, recoveries were observed in the preserved peptide glycerin at low-pH matrix when using the NOE, and recoveries below acceptance criteria were observed when using the LPS. To rule out the effect of the preservative, a second peptide in a tris buffer matrix at neutral pH was evaluated. The challenge was successfully recovered in the neutral pH peptide drug product using either NOE or LPS, although the NOE was less variable (Figure 6).

A preserved peptide solution at approximately pH 4.0 (Pep1) was observed to have lost the commercial endotoxin challenge in 4 days. Reduction to around the lower acceptance criteria was observed when using a NOE, indicating the purified LPS is more susceptible to matrix pH affects. A similar preserved peptide buffered at pH 7.4 with Tris (Pep2) suitably recovered both NOE and LPS challenges.
The same endotoxin stock suspension preparation was used repeatedly at the originating site laboratory, with a quantitation control performed prior to each experiment. When stored under refrigeration as a liquid suspension in a polystyrene container, the stock averaged 4.2 × 106 EU/mL (%CV = 26%) over 398 days to date. The observed variation is susceptible to the assay y-intercept variability. Y-intercept variability in endotoxin challenge studies could potentially be mitigated by analyzing subsequent time points to the initial standard curve. The choice to use the same E. coli strain as used to prepare the internal platform commercial CSE is one of assured specificity, and for the same reasons that the RSE E. coli was chosen as a model endotoxin.
Conclusion
The NOE challenges were successfully recovered in the matrices of interest, and the LER effect could not be reproduced for citrate and phosphate buffer systems containing polysorbate. Therefore, the respective verified test methods are capable of detecting potential endotoxin in citrate, histidine/glycine, phosphate, and sodium acetate matrices containing polysorbate, and are not affected by time, typical protein concentration, refrigerated or frozen storage conditions, process product-contact materials, or LAL test reagent supplier. Recovery was also demonstrated using an orthogonal non-LAL method. No evidence was found that indicates the LER phenomena posed a risk of failing to detect endotoxin during end-product testing by compendia methods. Based on our findings, the harmonized compendia bacterial endotoxin chapters should continue to be used for routine endotoxins testing.
It is proposed here that a NOE liquid stock endotoxin preparation is as stable, representative, and suitable—or better—than an LPS. The E. coli endotoxin strains specific to a supplier's CSE/LAL are publicly available, can be used to maintain specificity to supplier CSE, and fit the use of a model organism for endotoxin challenge studies.
Conflict of Interest Declaration
The authors declare that they have no competing interests.
Acknowledgements
Dr. Dayue Chen, Dr. James Cooper, Dr. Michael DeFelippis.
- © PDA, Inc. 2014




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}