
Abstract
Extracts of plastic packaging, manufacturing, and delivery systems (or their materials of construction) are analyzed by chromatographic methods to establish the system's extractables profile. The testing strategy consists of multiple orthogonal chromatographic methods, for example, gas and liquid chromatography with multiple detection strategies. Although this orthogonal testing strategy is comprehensive, it is not necessarily complete and members of the extractables profile can elude detection and/or accurate identification/quantification. Because the chromatographic methods rarely indicate that some extractables have been missed, another means of assessing the completeness of the profiling activity must be established.
If the extracts are aqueous and contain no organic additives (e.g., pH buffers), then they can be analyzed for their total organic carbon content (TOC). Additionally, the TOC of an extract can be calculated based on the extractables revealed by the screening analyses. The measured and calculated TOC can be reconciled to establish the completeness and accuracy of the extractables profile. If the reconciliation is poor, then the profile is either incomplete or inaccurate and additional testing is needed to establish the complete and accurate profile.
Ten test materials and components of systems were extracted and their extracts characterized for organic extractables using typical screening procedures. Measured and calculated TOC was reconciled to establish the completeness of the revealed extractables profile. When the TOC reconciliation was incomplete, the profiling was augmented with additional analytical testing to reveal the missing members of the organic extractables profile. This process is illustrated via two case studies involving aqueous extracts of sterile filters.
LAY ABSTRACT: Plastic materials and systems used to manufacture, contain, store, and deliver pharmaceutical products are extracted and the extracts analyzed to establish the materials' (or systems') organic extractables profile. Such testing typically consists of multiple chromatographic approaches whose differences help to ensure that all organic extractables are revealed, measured, and identified. Nevertheless, this rigorous screening process is not infallible and certain organic extractables may elude detection. If the extraction medium is aqueous, the process of total organic carbon (TOC) reconciliation is proposed as a means of establishing when some organic extractables elude detection. In the reconciliation, the TOC of the extracts is both directly measured and calculated from the chromatographic data. The measured and calculated TOC is compared (or reconciled), and the degree of reconciliation is an indication of the completeness and accuracy of the organic extractables profiling. If the reconciliation is poor, then the extractables profile is either incomplete or inaccurate and additional testing must be performed to establish the complete and accurate profile. This article demonstrates the TOC reconciliation process by considering aqueous extracts of 10 different test articles. Incomplete reconciliations were augmented with additional testing to produce a more complete TOC reconciliation.
- Extractables
- Total organic carbon (TOC)
- Reconciliation
- Plastics
- Polymers
- Medical devices
- Packaging systems
- Manufacturing systems
Introduction
During their manufacturing, storage, and administration, pharmaceutical drug products come in contact with plastic systems designed to accomplish these operations. For example, the drug product may contact plastic materials present in the manufacturing suite (e.g., mixing tanks, bioreactors, tangential flow filter systems, sterile and non-sterile filters, filling lines, etc.) during the manufacturing process. After manufacturing but before use, the drug products may be stored in a plastic packaging system. Lastly, during use the drug product may be administered via a medical device that has plastic components. Although these systems are constructed with plastic materials and by processes that seek to minimize the interactions between them and the drug product, neither truly inert materials and systems nor truly benign contact conditions exist, and interactions with potential product quality impact are the rule rather than the exception.
One type of interaction that can occur between a drug product and a material that it contacts is the transport of chemical entities out of the material and into the product. The accumulation of such plastic-related substances in the finished drug product is of concern due to the impact that such substances could have on the finished drug product's suitability for use. It is both necessary and mandatory that the extent of such an interaction be ascertained and that it be established that the impact of the interaction is within acceptable limits.
One means of ascertaining the potential extent of the interaction is to characterize the plastic system (and/or its materials of construction) for extractable substances. Extracts of plastic packaging, manufacturing, and delivery systems (and/or their materials of construction) are generated and then analyzed (screened) by chromatographic methods to establish their extractables profile. The testing strategy involved in characterizing the extracts for their organic extractables typically consists of orthogonal chromatographic methods including gas chromatography with headspace sampling and both mass spectrometric and flame ionization detection (HS-GC/FID-MS, volatile compounds), gas chromatography with flame ionization and mass spectrometric detection (GC/FID/MS, semi-volatile compounds) and liquid chromatography with both ultraviolet absorption and mass spectrometric detection (LC/UV/MS, non-volatile compounds) (1⇓⇓⇓⇓⇓–7). Although this orthogonal testing strategy is comprehensive, it is not necessarily complete and members of a test article's extractables profile can elude detection and/or accurate identification/quantification by the screening methods. Because the chromatographic methods rarely provide an indication that some extractables have been missed, another means of assessing the completeness of the profiling activity must be established. For example, Thibon and Creasey reported the results of using total organic carbon (TOC) as a surrogate for the total amount of organic extractables extracted from materials used in single-use biopharmaceutical manufacturing systems and concluded that “the results demonstrated the need to look for a wide variety of compounds some of which are not currently detected by conventional techniques” (8).
It stands to reason that it is inefficient to devise and routinely apply a screening process for organic extractables that includes all the many diverse tests necessary to make screening a truly comprehensive process. In many cases, the standard, and well-recognized, primary screening methods can and will reveal all the essential members of a test material's extractables profile. In such cases, having additional secondary test methods will not produce valuable additional information. Rather, it is potentially more efficient to have a screening process that is largely comprehensive and then have a means of establishing those hopefully few circumstances where the screening is not comprehensive enough and where additional testing is required. If the extracts are aqueous and contain no organic additives (e.g., pH buffers or solubilizing agents), they can be analyzed for their TOC content. Additionally, the TOC of an extract can be calculated as the sum, for all organic extractables measured by the chromatographic analyses, of the individual products of the extractable's measured concentration in the extract and its carbon fraction. The measured and calculated TOC can be reconciled to establish the completeness and accuracy of the extractables profile. If the reconciliation is poor, then this is an indication that the profile is either incomplete or inaccurate and that additional testing must be performed to ascertain the complete and accurate profile.
This article summarizes the activity of screening the aqueous extracts of 10 test articles for organic extractables using typical chromatographic methods. Measured versus calculated TOC is used to establish those cases among the 10 test articles wherein the screening methods failed to reveal a significant portion of the extractables profile. In those cases, the screening methods were augmented with additional analytical testing, ultimately producing a more complete extractables profile. Such a process is illustrated in two case studies involving the extractables profiling of filters that could be used in the manufacturing of pharmaceutical products. It is noted that the TOC reconciliation process is specifically relevant to those extractables that are present in a specific extract. Extractables that may be present in a test article but not extracted by the medium used in a particular study are clearly not addressed by the TOC reconciliation. The TOC reconciliation is a concept that addresses the completeness with which a specific extract has been characterized and is not a concept that necessarily addresses whether an individual extract reflects a test article's complete extractable profile.
Experimental
Test Articles and Their Extraction
The test articles used in this study were plastic components of manufacturing or packaging systems. A generic description of these test articles, indicating their nature and essential materials of construction, is provided in Table I.
General Description of the Test Articles Examined in this Study
The test articles were extracted using aqueous media that included low-pH solutions (pH ≈ 3), higher pH buffers (pH ≈ 9), and water (at an ambient pH of 6). The test articles were extracted by a methodology appropriate for their nature by a static process. For example, the test articles that could be filled were filled with the extracting medium and then stored at a defined temperature for a defined period of time. Test articles that could not be filled were typically placed in inert extraction vessels and contacted with the extracting solution at a defined temperature for a defined period of time. For the purpose of this paper the exact details of the extraction processes are largely irrelevant and thus they are not reported herein. Appropriate extraction blanks were also generated and tested.
Chromatographic Screening of Extracts
The extracts and associated extraction blanks were screened for organic extractables using the three analytical approaches discussed previously. While the exact methodologies utilized by various laboratories may vary somewhat, the operating conditions and techniques used in the methods employed in this study are consistent with and comparable to those methods that are generally recognized as being necessary and appropriate for organic extractables profiling, including
Headspace gas chromatography with both flame ionization and mass spectrometric detection (HS-GC/FID-MS)
Gas chromatography (direct injection) with FID and MS detection (GC/FID-MS)
High-pressure liquid chromatography with both UV and mass spectrometric detection (LC/UV/MS)
Considering the GC/FID-MS method, the separation was accomplished with a capillary column (60 m × 0.25 mm) containing a non-polar, mixed phenyl/dimethylsiloxane stationary phase (0.25 μm) and an oven program that extended from 40 to 300 °C. The mass detector was operated in the electron impact mode over a mass range of 35 to 500 daltons. The aqueous extracts were prepared for the GC analysis by solvent exchange with methylene chloride and evaporative concentration of the resulting methylene chloride samples. The pH of the extracts was adjusted both high and low during the solvent exchange process to ensure that acidic and basic extractables effectively partitioned into the organic phase. Concentrated solvent-exchanged samples were analyzed “as is” and after derivatization to form trimethylsilyl derivatives. The column effluent was split so that the FID and MS detection could be accomplished simultaneously in a single sample injection.
Considering the LC/UV/MS method, the reversed phase separation was accomplished with a column (30 mm × 4.6 mm) containing an appropriate C18 stationary phase (2.5 μm particles) and a mobile phase gradient from 5 to 100% acetonitrile (the aqueous phase contained 10 mM ammonium acetate). Data from the UV detector was collected at four wavelengths (210, 230, 250, and 270 nm) and MS electrospray ionization (ESI) spectra were obtained in both the positive and negative ion mode over a mass range of 100 to 1500 daltons.
TOC Measurements
The TOC measurements were performed with appropriate analytical instrumentation. The analytical process involved acidifying the sample to a pH of 2 or less, followed by sparging the sample with nitrogen gas at a temperature of 70 °C to remove the inorganic carbon. The temperature is then increased to between 90 and 98 °C and sodium persulfate is added to the sample. Organic carbon in the sample reacts to form CO2, which is then detected by a nondespersive infrared detector. All TOC measurements were performed consistent with the system suitability expectations in USP <643> (9) (response efficiency not less than 85% and not more than 115%). However, because the TOC in the extracts is higher than in purified water, the system suitability tests were performed at higher TOC levels than the 0.5 mg/L specified in <643>.
TOC Reconciliation Calculation
The TOC reconciliation is calculated as follows:
The TOC concentration of an individual extractable (TOCe) is calculated as the product of the extractables's measured concentration in the extract (Cm) and the extractable's carbon fraction (as derived from the extractable's empirical formula, Cf) per eq 1.

The calculated TOC for the entire extractables profile (TOCcalc) is obtained by adding up the individual TOC concentrations (TOCe) for all extractables per eq 2.
The TOC reconciliation (TOCrec), expressed as a percentage, is calculated as the ratio of the calculated TOC for the entire profile (TOCcalc) and the TOC measured for the extract (TOCmeas) per eq 3.


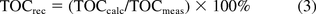
Case Studies
The specific test article that was the subject of the TOC reconciliation Case Study #1 was a 0.2 μm sterile capsule filter (2 ft2) that consisted of a Nylon 6.6 filter membrane and a polyester shell and support (Article 9 in Table I). The specific test article that was the subject of Case Study #2 was a 0.2 μm sterile capsule filter (10”) that consisted of a polyvinylidene fluoride (PVDF) filter membrane with polypropylene support, housing, and end caps (Article #10 in Table I). This filter was sterilized using gamma irradiation.
For Case Study #1, replicate filters were extracted by filling them with either a pH 3 salt mixture or a pH 9 phosphate buffer and storing the filled filters at 40 °C for 24 h, slightly exaggerating the filter's typical conditions of use. For Case Study #2, replicate filters were extracted by filling them with either a pH 5 or a pH 9 phosphate buffer and storing the filled filters at 25 °C for 2 h. For both studies, the filled filters were shaken during extraction and after the allotted time were cooled to ambient temperature and emptied. The emptied fill solution was the extract; extractions blanks were unused portions of the extraction solution.
The extracts and extraction blanks were characterized via the previously described screening test methods. Additionally, the extracts and blanks were analyzed by three auxiliary methods to address deficiencies in the screening methods. Ion exclusion chromatography (IEC) with UV detection was used to screen the extracts for low molecular weight organic acids. Separation was accomplished on a Biorad Aminex HPX-87H ion exclusion column with a 0.005 M sulfuric acid mobile phase (flow rate = 0.7 mL/min). UV detection was accomplished at a wavelength of 210 nm. IEC with UV detection was used to screen the extracts for low molecular weight gycols. Separation was accomplished on a Biorad Aminex HPX-87H ion exclusion column with a 0.1% formic acid mobile phase (flow rate = 0.7 mL/min). The targeted analytes were detected and quantified via MS ESI positive ion–extracted ion chromatograms. A reversed phase separation was coupled with a charged aerosol detector (CAD) in order to quantify extractables that could not be quantified from the LC screening runs due to lack of a reference standard material and/or suitable surrogate. The separation was accomplished with an HP Acclaim 120 C18 column (100 × 3 mm, 2.2 μm particles) and a mobile phase gradient formed from water (0.1% formic acid) and acetonitrile (1% formic acid) with a combined flow rate of 1.0 mL/min. An equal but opposite proportion gradient was added post-column to the column effluent to enable the introduction of an isocratic solution to the CAD detector. A Thermo Corona Ultra RS CAD detector was used consistent with the manufacturer's recommendations.
Results and Discussion
General Discussion
Figure 1 illustrates the TOC reconciliations obtained when aqueous extracts of various test materials were analyzed for TOC and screened for organic extractables by the GC, HS-GC, and LC screening methods described previously. The percentage of the TOC reconciled by the extractables measured by each technique is shown by the appropriate color code, and the percentage of the TOC that was not reconciled by the extractables uncovered and quantified by these methods was labeled as “unknown” and is likewise indicated. Because some extractables can be detected by multiple screening methods, it is possible for a compound to be counted multiple times in the reconciliation. To prevent this multiple counting, the largest concentration obtained for a particular compound by any test method was the number that was used for that compound in the TOC reconciliation.

TOC reconciliation results for the 10 test articles. The shaded areas indicate that portion of the TOC that was reconciled by the organic extractables uncovered by the various chromatographic techniques. The area shaded in purple is thus that portion of the TOC that is due to organic extractables that were not uncovered by the chromatographic screening methods. While the screening methods were well suited for the extracts of the bag materials, the methods were less comprehensive for characterizing filter extracts.
The TOC for materials 5, 6, and 8 was effectively reconciled by this process. In the case of materials 5 (pH 3), 6 (pH 3) and 8 (both pH values), the TOC reconciliation was 80% or higher, suggesting that a majority of the extractables associated with these test articles were revealed, correctly identified, and accurately quantified. In the case of materials 5 and 6 at pH 9, a TOC reconciliation of greater than 100% was obtained. Although this outcome may seem to be more unusual than a TOC reconciliation that is lower than 100%, it is the case that such high results could be the natural and logical outcome of individual extractables whose concentrations are overestimated somewhat by the screening methods.
It is noteworthy that the TOC reconciliations were best for those test articles that are traditionally the target of extractables assessment, such as bags and other packaging components. This is the case because extractables from such test articles are well known and the chromatographic methods were devised and optimized to be responsive to these types of materials (e.g., polyvinyl chloride [PVC], polyolefins, elastomers) and their associated extractables (antioxidants, plasticizers, additives and the like). The TOC reconciliations were incomplete, and generally less than 20%, for the less traditional test articles such as filters used in manufacturing systems. Such an outcome occurs because extractables from such test articles are less well known and tend to be chemically different than the extractables from bags and packaging systems. For example, filter membranes whose backbone polymer may be hydrophobic can be made more hydrophilic by coating them with wetting agents based on polyethylene glycols. Such agents, which may be readily extracted from the filters, escape the screening methods described previously for several reasons, including
lack of volatility (making HS-GC and GC poor choices for their measurement)
high water solubility (meaning that they might not be retained on the typical reversed phase C18 stationary phases used in traditional LC methods and that they may not partition favorably in the solvent switching/analyte concentrating sample preparations steps used prior to GC analysis)
UV transparency (thereby producing little if any LC/UV response)
poor ionization (thereby producing little if any LC/MS response)
high molecular weight (potentially falling outside the operating range of a typical LC/MS detector)
The solution to incomplete TOC reconciliations is clear and inescapable; additional analytical methods must be employed to find those extractables that eluded detection or accurate identification and quantitation by the screening methods. Oftentimes, the nature of the test material itself will suggest possible missing extractables, and this knowledge can be used to effectively identify appropriate supplementary analytical methods.
Case Studies; Improving the TOC Reconciliation Obtained for Filter Extracts
Figure 2 more clearly illustrates the TOC reconciliation process used in the two case studies. In both case studies, the screening analyses fail to reveal a sufficient number of extractables, resulting in an extract TOC that was poorly reconciled and suggesting that the screening methods need to be augmented in order to reveal the test article's complete extractables profile.

TOC reconciliation results for the two case studies. In Case Study #1, use of LC/CAD to quantitate the Nylon 6.6 oligomers effectively closed the TOC reconciliation gap. In Case Study #2, use of ion exclusion LC/UV to quantify low molecular weight acids and glycols effectively closed the TOC reconciliation gap.
Figures 3 through 7 are representative chromatograms obtained by the GC and LC screening analysis of the aqueous extracts of the case study #1 filter. HS-GC chromatograms obtained as part of the screening activity (Figure 3) revealed that presence of isopropyl alcohol (IPA) at readily measurable levels in the extracts. For this reason, the extracts were re-analyzed by HS-GC for the specific purpose of quantifying IPA. The HS-GC analysis also revealed the presence of lesser quantities of tetrahydrofuran and 2-ethyl-1-hexanol in the extracts. GC chromatograms for both underivatized and derivatized concentrates of the filter extracts are shown in Figures 4 and 5 and confirmed the presence of phenol and 2-ethyl-1-hexanol in the extracts. Additionally, the GC analyses revealed 2-ethyl-1-hexanoic acid and 2-phenyl-2-propanol in the extracts.

Headspace GC/MS chromatograms for the extracts of the Case Study #1 filter. The major extractables revealed by the HS-GC testing included isopropyl alcohol (IPA), tetrahydrofuran (THF) and 2-ethyl-1-hexanol (EHO).

GC/FID chromatograms, underivatized sample preparation of the extracts for the Case Study #1 filter. The predominant extractables revealed by this analysis included phenol, 2-ethyl-1-hexanol (EHO), and 2-phenyl-2-propanol (PP).

GC/FID chromatograms, derivatized sample preparation of the extracts for the Case Study #1 filter. The predominant extractables revealed by this analysis included phenol and 2-ethyl-1-hexanol, measured as their TMS derivatives.
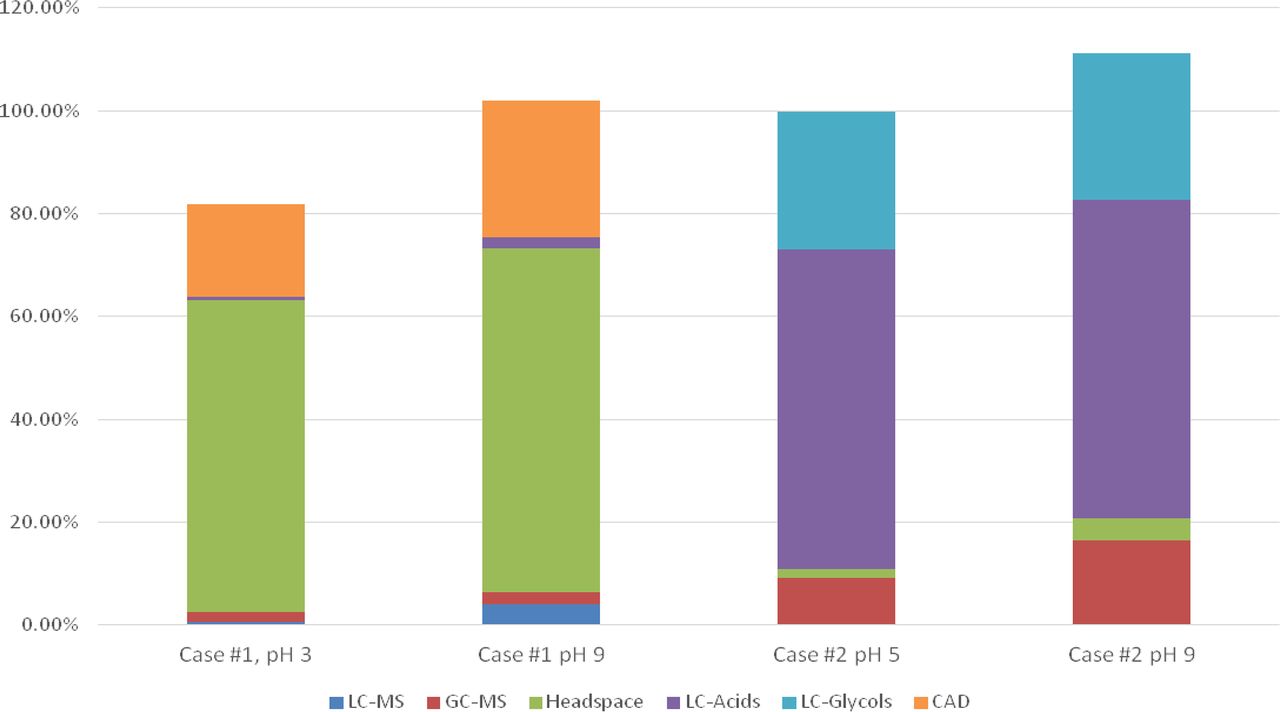
LC/MS chromatograms, negative ion detection mode, are illustrated in Figure 6. The two predominant peaks in these chromatograms were ascribed to Nylon 6.6 oligomers, specifically the tetramer and the hexamer. The LC/MS positive ion chromatograms essentially revealed the same two extractables. LC/UV chromatograms were essentially devoid of recognizable peaks. The identification of nylon oligomers as major filter extractables is consistent with previous reports that document linear and cyclic oligomeric extractables that are based on nylon's repeating monomers (10, 11).

LC/MS base peak chromatogram, negative ion mode, of the filter extracts (Case Study #1). While the method was able to establish the presence extractables in the extracts, and suggested that several of the extractables were Nylon 6.6 oligomers, these identities could not be confirmed and the extractables could not be quantified by this method due to lack of appropriate reference materials.
While the LC/MS data was sufficient to suggest probable identities for these extractables, lack of an authentic reference material or otherwise reliable surrogate standard prevented them from being quantified by this approach.
The levels of the identified and quantifiable aqueous extractables for the case study filter were used to calculate a TOC reconciliation, the results of which are shown in Table II. Cumulatively, the identified and quantified organic extractables accounted for approximately 63% of the pH 3 extract's TOC and approximately 73% of the pH 9 extract's TOC, with the major contributor to the TOC being IPA. As this reconciliation indicates that the aqueous extracts contained organic extractables that were not revealed and/or quantified by the screening methods, it was appropriate to search for and measure those missing extractables.
Case Study #1, Chemical Characterization of the Test Article Extract
Un-reconciled TOC can either be associated with extractables that were not detected in the screening methods (missing extractables) or that were not properly identified or quantified by the screening methods (unfinished extractables). Which of these circumstances is the case in a given study dictates the appropriate strategy to fill the TOC gap. In the circumstances of this case study, both situations exist. Firstly, there are unfinished extractables (the nylon oligomers) that were detected but not quantified by the screening methods. Clearly, once these unfinished extractables are quantified, the test results can be factored into the TOC reconciliation and the reconciliation can be improved. However, it was anticipated that consideration of these unfinished extractables alone would not fully close the TOC gap, as the data suggests that there remains one (or more) missing extractables that were not captured by the screening methods. In such a circumstance, new methods must be employed to find these missing extractables. The question is, where should one look?
Considering the quantitation of the of the nylon oligomers, the issue is not a lack of chromatographic response but rather the lack of a means to correlate the magnitude of the analytical response to the concentration of the oligomers in the extract. Two potential solutions to this issue are readily apparent:
Authentic reference standards can be generated (for example, by either synthesis or isolation/purification) and qualified, or
A detection mechanism with a universal response can be employed with one or more surrogate standards
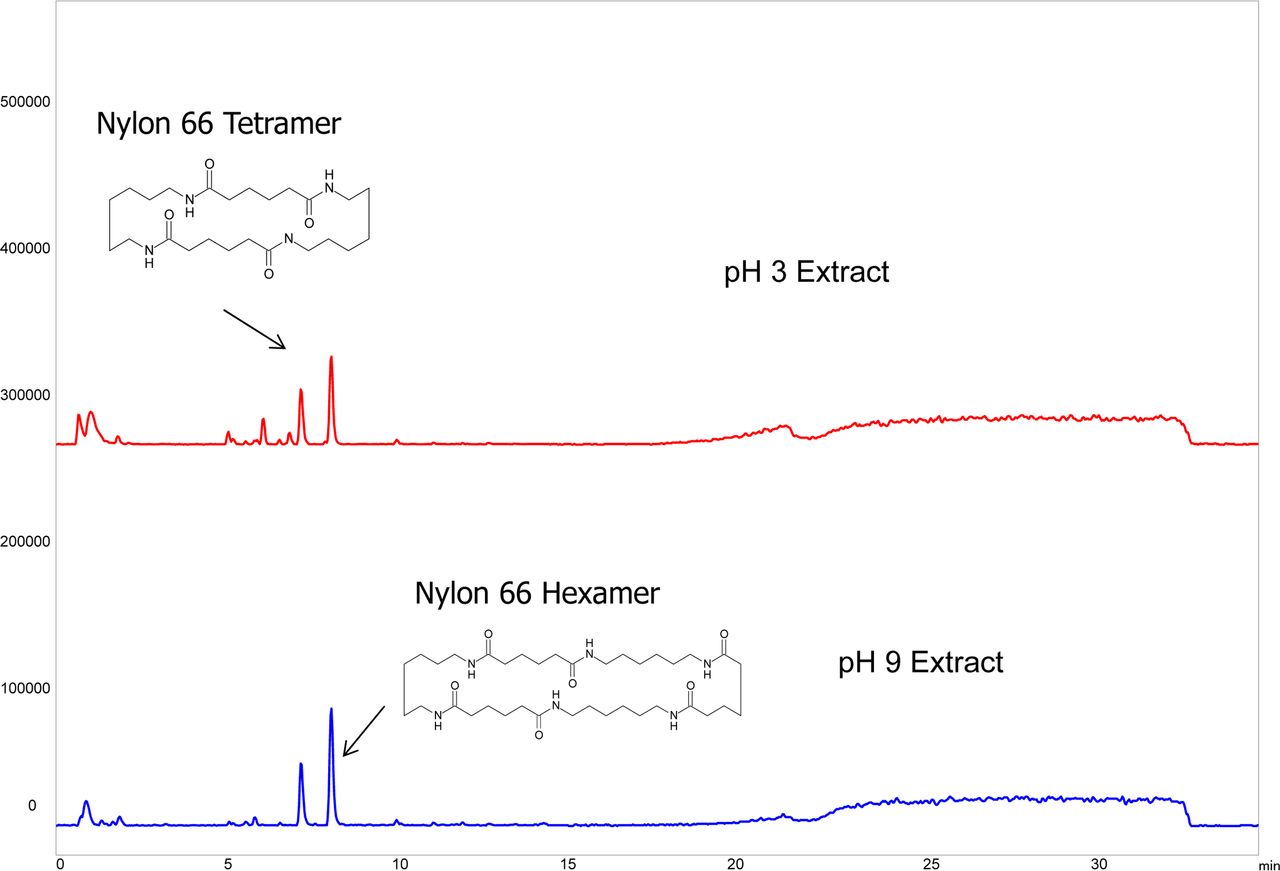
In this case study, the latter approach was taken. To wit, a CAD was coupled with LC to quantitate the nylon oligomers. Such a detection approach, which is based on the evaporation of the column efficient and charging of the resultant particles, has been utilized in extractables studies (12) and is well suited for the higher molecular weight, relatively non-volatile oligomers. As shown in Figure 7, both oligomers produced adequate LC/CAD responses for quantitation and thus the oligomers were quantitated by this method.

LC/CAD chromatogram of the pH 3 extract illustrating the chromatographic peaks associated with the Nylon 6.6 oligomers (Case Study #1 filter). Both oligomers produced strong chromatographic signals that could be used to quantify the oligomers via the use of appropriate surrogate standards, taking advantage of the CAD detector's universal response.
Considering the case of the missing extractables, one examines the existing data to gain insight into the nature of the missing extractables so that the appropriate means of finding the missing extractables can be effectively and efficiently established and implemented. For example, material extracts can be tested by methods that provide little or no information with respect to individual extractables but that establish the general chemistry properties of the extractables. For instance, measuring the pH of an extract and comparing it to the pH of an extraction blank can be used to establish the presence of acidic or basic extractables. Similarly, measuring the UV absorption spectrum of an extract can be useful in establishing the general functionality of the extractables (e.g., aromatics versus aliphatics). In the case of the test filter, unbuffered extracts of the filter experienced a discernible pH drop (versus the extraction blank), suggesting that the suite of extractables included acids. In the case of UV absorbance, the UV spectra of the extracts were consistent with the types of extractables that had already been addressed by the screening methods and thus provided no insight into the identities of any undetected extractables.
Low molecular weight organic acids (e.g., acetic, formic, propionic, and adipic acids) can be separated by IEC and detected by direct UV detection at concentrations that are low enough for extractables quantitation. Thus, this approach was used to screen the aqueous extracts for these types of extractables, and formic acid was found to be present in both the low- and high-pH extracts.
Armed with the oligomer and organic acids data, the TOC reconciliation was recalculated. So doing decreased the TOC gap considerably, producing a 82% reconciliation in the pH 3 extract and a 102% reconciliation in the pH 9 extract. For the purpose of this case study, including the oligomer and organic acid data to extractables profile, completed the profiling process.
A similar, but more dramatic, example of the use of supporting methods to establish the complete extractables profile of a test article involved Case Study #2. In this study, the screening methods were largely unsuccessful in revealing the organic extractables; as noted in Table III, the TOC reconciliations obtained based on the results of the screening methods was less than 30%. HS-GC and LC chromatograms were largely devoid of peaks that could be ascribed to extractables. While the GC chromatograms contained numerous peaks associated with organic extractables, the levels of the all the individual extractables were low and no single extractable was present in the extract at a concentration greater than 0.9 mg/L.
Case Study #2, Chemical Characterization of the Test Article Extract
Two pieces of information suggested a logical approach for discovering the remaining extractables. Firstly, it was observed that the pH of the filter extracts had decreased versus the pH of the extraction blanks, suggesting that the extractables included organic acids. This possibility is further supported by the fact that the filters were sterilized by irradiation, as the generation of low molecular weight acids from irradiated plastics is well documented (13⇓⇓⇓–17). Thus it was anticipated that using the supporting assay for organic acids would reveal additional extractables. Secondly, it is noted that the filter membrane was hydrophilic PVDF. Previous extractables studies performed on such filters have reported extractable glycols in readily measurable quantities, where the glycols are specifically related to the surface modification required to render the PVDF material hydrophilic (18, 19). Thus it was further anticipated that using the supporting assay for low molecular weight glycols would also reveal extractables.
As shown in Table III, these expectations were realized when the extracts were tested using the supporting methods. Readily measurable quantities of acetic and formic acids and several low molecular weight glycols were revealed by the supporting methods. These results, coupled with the result of the screening methods, produced TOC reconciliations (105–128%) that suggest that the entire analytical process effectively delineated the extractables profile of this test article.
Utilizing TOC Reconciliation To Judge the Completeness of Extractables Profiling
Once the TOC reconciliation concept has been developed, the inevitable question follows: What range in TOC reconciliation values represents a complete extractables profile? In asking this question, one is looking for a response that sounds something like “If the TOC reconciliation is between 70 and 130%, then the extractables profile has been effectively and properly delineated”. There are two aspects of the TOC reconciliation and safety assessment processes that have a direct impact on the proper consideration of this question: analytical uncertainty and the safety impact of the unreconciled TOC. Considering the former, it is recognized that the calculated TOC reflects not only the sum of the carbon contributions of every individual extractable but also the analytical imprecision in each result for each extractable. Thus, the imprecision of the TOC reconciliation does not reflect the imprecision in the quantitation of an individual extractable but rather the aggregate imprecision in the quantitation of each and every extractable. For an extractables profile that consists of five compounds, each of which is quantitated to a precision of ±10% relative standard deviation, the imprecision of the calculated TOC is on the order of ±25%. Thus TOC reconciliation is, in most cases, insufficiently precise to lend itself to generalization in terms of “what range of TOC reconciliations means that the profiling activity is done?” Rather, the TOC reconciliation is a semi-quantitative tool that is most effectively used to establish those cases when the extractables profiling clearly is not complete or is inaccurate. In this context it is suggested that
If the TOC reconciliation is less than 50%, this is a clear indication that the extractables profile has not been fully delineated.
If the TOC reconciliation is between 75% and 125%, this is a clear indication that the extractables profile has likely been effectively delineated.
If the TOC reconciliation is greater than 150%, this is a clear indication that the extractables profile has not been effectively delineated. Such a result typically would suggest that the individual extractables concentrations have been overestimated.
The discussion of analytical imprecision notwithstanding, a more significant issue may be the difference between percent reconciliation and the amount of TOC that is un-reconciled. In the end, the safety or product quality impact of undiscovered extractables (as potential leachables) lies not in the percentage of TOC that is un-reconciled but rather the amount of TOC that is un-reconciled. A numerical example illustrates this point. In this example, two components are candidates for use in a packaging system. To facilitate the selection of the best candidate, an extractables study is performed on both candidates. In this study, the two components are extracted in the same way and the extracts are tested in an analogous manner. For component 1, the measured TOC of the extract was 10 mg/L and the TOC reconciliation was 90% (meaning 1 mg/L TOC was not reconciled). For component #2, the measured TOC of the extract was 2 mg/L but the TOC reconciliation was only 60% (meaning 0.8 mg/L TOC was not reconciled). Thus the unknown safety risk, linked to the amount of unreconciled TOC, is greater for the case with the higher TOC reconciliation. From a safety risk perspective, the 60% reconciliation of the smaller TOC is better (less safety risk due to unknown extractable) than the safety risk of the 90% reconciliation of the larger TOC.
It is beyond the scope of this article to provide a recommendation for how one establishes the safety risk of un-reconciled TOC. Nevertheless, the following mathematical example may serve to illustrate one such approach. Consider a container used to package a once-a-day dose of a particular drug product. The fill volume of the container is 10 mL. The product is dosed for a period of 1 week to address an acute health issue. The TOC of the extract obtained in an extractables simulation study (extraction conditions closely simulate the chemical nature of the drug product and the temperature and duration of contact between the drug product and the packaging) is 4 mg/L. The TOC reconciliation was 85%. The question is, “what is the safety impact of the un-reconciled TOC?”
To address this question, one notes that the magnitude of the un-reconciled TOC is 4 mg/L × 0.15 × 0.01 L = 0.006 mg or 6 μg carbon. If one makes the generalization that the carbon content of a “typical” extractable is 60%, then the un-reconciled corresponds to 6 μg/0.6 = 10 μg of un-reconciled extractable. It is clear that such a calculation is only an approximation, as the carbon content of extractables can vary widely (for example, as low as 26.1% for acetic acid and as high as 94.1% for propylene glycol). Thus our original question can now be rephrased, “what is the safety impact of 10 μg/day of unknown extractables dosed to a patient in an acute therapy persisting for 7 days?” While it is beyond the scope of this article to answer this question, as this transcends the concepts of TOC reconciliation and represents an exercise in toxicological safety assessment, it is noted that such an assessment can be readily performed.
Conclusion
Characterization of plastic materials used in packaging, manufacturing, and delivery systems for pharmaceutical products for organic extractables involves generating and chemically analyzing an extract. It is the generally recognized standard practice that the analysis of extracts includes the use of chromatographic screening methods to surface, identify, and quantify the extractables. As powerful as the state-of-the-art screening methods are, they may not account for all organic extractables. For aqueous extracts, comparing the extract's measured level of TOC to a calculated TOC level (based on the identity and concentration of individually measured extractables) provides a means of determining the degree to which a test material's extractables profile has been elucidated via by the screening methods. If the reconciliation of measured and calculated TOC is incomplete, the extractables profile has not been fully elucidated and non-screening methods must be employed to account for the missing or incomplete extractables. In addition to reporting the TOC reconciliations obtained for 10 test materials, this manuscript contains two case studies wherein the screening methods did not produce an adequate reconciliation. The ramifications of the TOC gap were considered, and appropriate means were taken to close the TOC gap with testing and testing methods that targeted the suspected missing and known incomplete extractables. An LC/CAD method was used to quantify incomplete extractables (Nylon 6.6 oligomers) which had no corresponding reference standards. Additionally, LC/UV methods based on an ion exclusion separation were used to profile the extracts for targeted organic acids and glycols as missing extractables. In the case of hydrophilic filters that are made from intrinsically hydrophobic polymers, the wetting agents are particularly challenging to detect using conventional screening methods.
TOC reconciliations of 80–120% were obtained in two case studies by using the data from both the screening and profiling test methods, suggesting that the test material's extractable profile had been fully and effectively established by the screening and auxiliary testing.
Thus TOC reconciliation can be an effective tool to direct an analyst back into the laboratory to discover, identify, and quantify extractables that were not detected in screening methods. Analytical methods that target specific classes of extractables, such as those considered in this paper, can be used to accomplish these objectives.
Conflict of Interest Declaration
The authors declare that they have no competing interests.
- © PDA, Inc. 2014




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}