
Abstract
The risk mitigation of extractables and leachables presents significant challenges to regulators and drug manufacturers with respect to the development, as well as the lifecycle management, of drug products. A holistic program is proposed, using a science- and risk-based strategy for testing extractables and leachables from primary containers, drug delivery devices, and single-use systems for the manufacture of biotechnology products. The strategy adopts the principles and concepts from ICH Q9 and ICH Q8(R2). The strategy is phase-appropriate, progressing from extractables testing for material screening/selection/qualification through leachables testing of final products. The strategy is designed primarily to ensure patient safety and product quality of biotechnology products. The holistic program requires robust extraction studies using model solvents, with careful consideration of solvation effect, pH, ionic strength, temperature, and product-contact surface and duration. From a wide variety of process- and product-contact materials, such extraction studies have identified and quantified over 200 organic extractable compounds. The most commonly observed compounds were siloxanes, fatty acid amides, and methacrylates. Toxicology assessments were conducted on these compounds using risk-based decision analysis. Parenteral permitted daily exposure limits were derived, as appropriate, for the majority of these compounds. Analysis of the derived parenteral permitted daily exposure limits helped to establish action thresholds to target high-risk leachables in drug products on stability until expiry. Action thresholds serve to trigger quality investigations to determine potential product impact. The holistic program also evaluates the potential risk for immunogenicity. This approach for primary drug containers and delivery devices is also applicable to single-use systems when justified with a historical knowledge base and understanding of the manufacturing processes of biotechnology products.
LAY ABSTRACT: In the development of a drug product, careful consideration is given to impurities that may originate from manufacturing equipment, process components, and packaging materials. The majority of such impurities are common chemical additives used to improve the physicochemical properties of a wide range of plastic materials. Suppliers and drug manufacturers conduct studies to extract chemical additives from the plastic materials in order to screen and predict those that may leach into a drug product. In this context, the term extractables refers to a profile of extracted compounds observed in studies under harsh conditions. In contrast, the term leachables refers to those impurities that leach from the materials under real-use conditions and may be present in final drug products. The purpose of this article is to present a holistic approach that effectively minimizes the risk of leachables to patient safety and product quality.
- Extractables
- Leachables
- Safety concern threshold
- Action threshold
- Biotechnology products
- Single-use systems
Introduction
The development and manufacturing of a drug product are subject to regulations that manage and control the risk of impurities to patient safety and product quality. The International Conference on Harmonization (ICH) offers guidelines on pharmaceutical development (Q8) (1) and quality risk management (Q9) (2). In conjunction with these guidelines are also those on the general impurities in new drug substances (Q3A) (3) and in new drug products (Q3B) (4). The ICH also has guidelines specific for residual solvents (Q3C) (5), DNA-reactive (mutagenic) impurities (M7) (6), and elemental impurities (Q3D) (7). However, the biological/biotechnological products and the leachables in these products are not in the scope of ICH Q3A, Q3B, and Q3C. It is only in ICH Q3D and M7 guidelines that leachables are included as appropriate, namely, as elemental or genotoxic impurities. While there are currently more consistent practices for risk control or mitigation of leachables for orally inhaled nasal drug products (OINDPs), such practices vary widely for biotechnology products.
The suppliers of packaging materials and the manufacturers of drug product generate extractables profiles under experimental conditions using model solvents. However, it is the ultimate responsibility of the drug product manufacturer to ensure that leachables do not adversely affect patient safety or product quality. The Product Quality Research Institute (PQRI) has made a significant contribution towards best practices for extractables and leachables (E&Ls) assessments in OINDPs (8, 9) and is currently focusing on the parenteral and ophthalmic drug products (PODPs) to classify the leachables into genotoxicants, irritants, sensitizers, and other toxicants, with a proposed threshold for each class (10, 11). Subject to the validation of this classification scheme and a dataset large enough for statistical analysis, leachables will be considered for qualification on the basis of their respective thresholds. However, beyond safety considerations, biotechnology products require additional considerations of the product quality attributes. Biotechnology products are more susceptible to structural modifications than chemically synthesized drug products, primarily due to their large molecular weights, complex structures, and abundance of binding sites on their surfaces (11, 12). Structural modifications may alter product quality, safety, and/or efficacy. Therefore, risk mitigation of E&Ls requires a holistic understanding of the process- and product-contact materials upstream and downstream of the purification processes, as well as recommended storage conditions in primary drug containers until expiry.
The purpose of this paper is to share the knowledge base on biotechnology products to advance best practices for risk management and control of leachables.
Materials and Methods
Materials
A wide variety of primary container closure systems, on-body devices, bulk drug storage containers, and disposable single-use bioprocess materials that come into direct or indirect contact with drug substance or drug product have been studied. Extraction studies were commonly conducted with assembled systems, and the identified extractable compounds were linked to the critical components of the systems, as appropriate, by confirmatory studies. The materials of construction for the components, as well as the testing per USP and International Organization for Standardization (ISO) requirements, were verified against the vendor data packages. The common polymers of the test materials included polyethylene (PE), polypropylene (PP), polycarbonate (PC), fluorinated ethylene-propylene FEP), and ethylene tetrafluoroethylene (ETFE). The common elastomers for the test materials included bromobutyl elastomer and chlorobutyl isoprene blend. The single-use bioprocess systems often used multi-layer films constructed of polyethylene (PE), polyester, poly-vinylidene dichloride (PVDC), ethyl vinyl alcohol (EVOH), ethylene vinyl acetate (EVA) copolymer, polyamide (PA), and polyethylene terephthalate (PET). The accessories of the single-use bioprocess systems were also studied; these included tubings, connectors, and filter cartridges. The materials of construction for these accessories included polydimethylsiloxanes (PDMSs), polypropylene (PP), and polysulfone (PS).
Extraction Methods
Extraction studies were conducted with model solvents that mimicked the range of solvation effect, pH, process conditions, and ionic strength of the product formulations. The typical solvents included deionized water and an aqueous organic solvent. A mixture of 20% acetonitrile, 20% ethanol, and 60% water is capable of solubilizing more organic substances than water, is miscible in water, and compatible with liquid chromatography–mass spectrometry (LC-MS), high-performance liquid chromatography (HPLC), evaporative light-scattering detection (ELSD), and gas chromatography–mass spectrometry (GC-MS) analytical techniques. The percentage of acetonitrile/ethanol was used to mimic organic content (protein, excipients) of certain biopharmaceutical formulations. The extraction conditions represented the high end of the process applications and product storage conditions, including temperature, contact surface, extraction volume, contact duration, and sterilization methods of the test materials. The experimental conditions were appropriate for biotechnology drug products without impairing the function and integrity of the material contact surfaces.
The extracts were analyzed for elemental impurities by inductively coupled plasma mass spectrometry (ICP-MS) and for organic compounds using GC-MS, HPLC-UV, and HPLC-MS. This wide array of analytical techniques enabled the orthogonal approach necessary for the data analysis (see Results and Discussion). The analytical capabilities for discovering, identifying, and quantifying extractable compounds are shown in Table I. The compounds were identified against commercial reference standards and quantified as amounts per system or per assembly (e.g., prefilled syringe system or single-use bioprocess bag), thereby allowing for estimates of exposure per patient dose for later stages of clinical development.
Analytical Capabilities for Discovering, Identifying, and Quantifying Extractables and Leachables
Toxicology-based Risk Assessment Methods
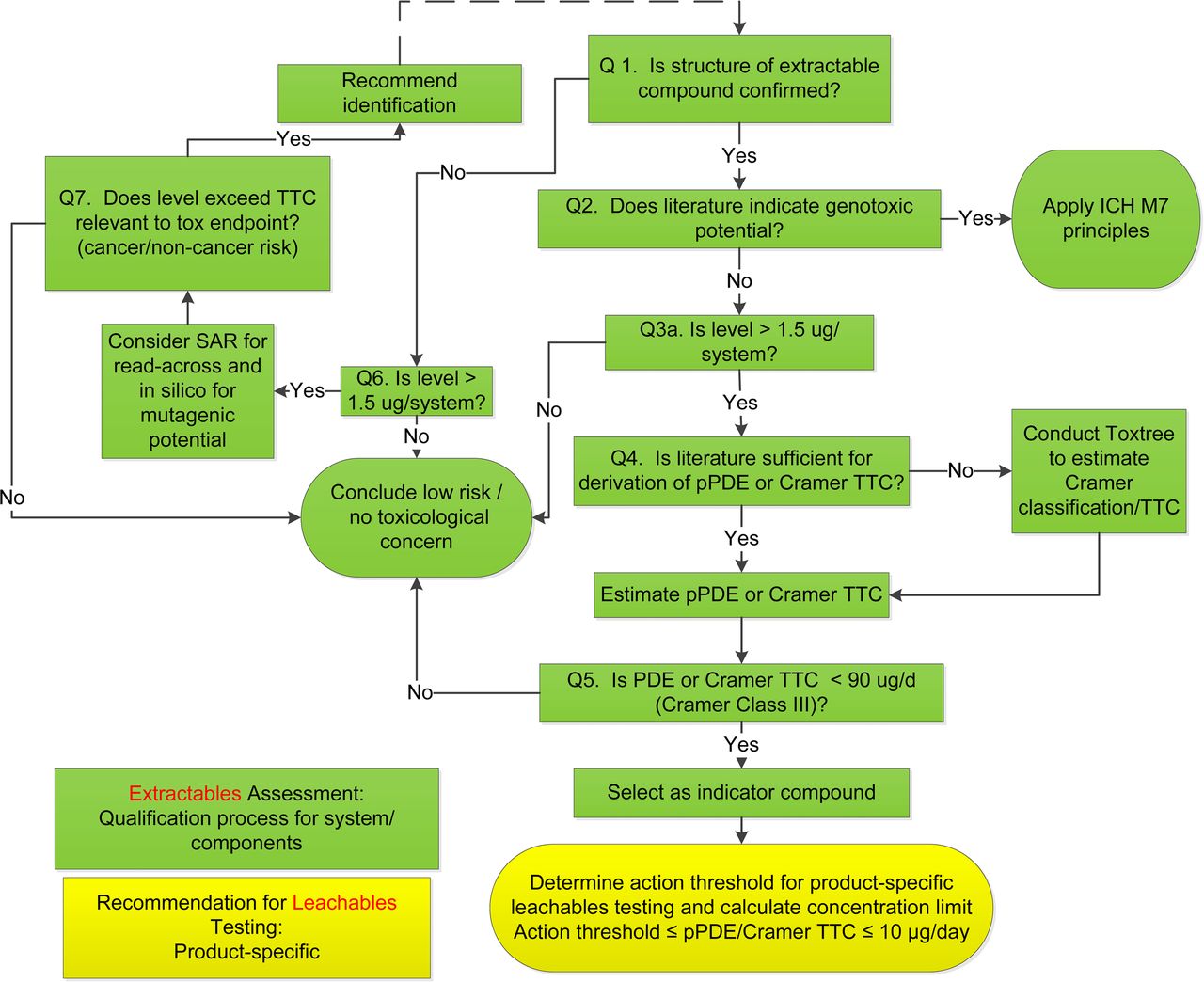
Literature searches were conducted on the identified organic compounds for toxicology data, supplemented with in silico analyses as necessary using Toxtree (13). The literature sources included, but were not limited to, the databases in ExPub, ToxNet, ECHA, Ovid, and SciFinder®. The toxicology assessments used a decision tree, as shown in Figure 1, for genotoxic risk (6, 14) and non-genotoxic risk (15⇓⇓⇓–19). After the risk of genotoxicity was excluded, parenteral permitted daily exposure (pPDE) values were derived following the principles and methods in ICH Q3C (5), with an additional modifying factor for bioavailability to account for parenteral exposure (see Results and Discussion).

Decision tree for science- and risk-based analysis of extractables and leachables profiles. A typical toxicology assessment of an extractables profile is conducted for the purposes of materials screening, selection, and qualification (steps colored in green). Based on the assessment, indicator compounds are selected to determine the action thresholds for leachables testing of final drug product on stability programs (step shown in yellow).
A typical review of the elemental impurities generally followed the safe levels established by the European Medicines Agency (EMEA) (20), ICH Q3D (7), and the Institute of Medicine (IOM) (21). The elemental impurities identified by ICP-MS were screened for metals of serious toxicity: arsenic (As), lead (Pb), cadmium (Cd), and mercury (Hg). ICH Q3D (7) and USP <232> have limits on these metals that require their detection at trace levels exceeding such limit (7).
As an example of inadvertent contamination, polystungstate was traced to the tungsten pins used for the forming process of syringe barrel. Because tungsten is known to interact and modify certain protein structures (22, 23), prefilled syringe components are subject to a limit on tungsten levels to mitigate such risk.
Product Impact Assessment Methods
The compounds identified in the extraction studies were evaluated for their potential for protein covalent interactions by electrophilic chemical mechanisms as reviewed by Aptula et al. (24), Enoch et al. (25), and others. The mechanisms searched included, but were not limited to, protein binding, Michael-type additions, Schiff base formation, acylation, nucleophilic aromatic substitution, and second-order nucleophilic aliphatic substitution. In addition to literature reviews, in silico screens were conducted with the Toxtree software program (13) with algorithms for structural alerts for protein binding (26) and Michael acceptors (27).
Results and Discussion
In the development of the holistic E&L program for biotechnology products, the principles and concepts of ICH Q9 (2) and ICH Q8(R2) (1) were incorporated. Leachables are inevitable when one considers all the polymeric surfaces that a drug substance or drug product comes into direct and indirect contact with throughout manufacturing, fill and finish, storage, and transportation (see Figure 2). Therefore the management and control of leachables need a thorough understanding of the potential sources and possible interaction that may alter the final drug product. Such understanding begins with supplier quality management and material screening, selection, and qualification. Extraction studies must be properly designed such that the extractable profiles from contact materials are predictive of leachables in the final drug product. This would include the appropriate model solvents to mimic the formulations for protein therapeutics. Whenever possible, extraction studies should be conducted with fully assembled components and systems that have undergone all processing steps such as washing, gamma irradiation, pressure, and autoclaving. Capturing such processing steps will avoid surprises of degradation products or unexpected leachables (28). When the processing steps are taken into account, ample examples (29, 30) show that leachables can be better predicted and the unexpected leachables become a small subset of extractables.

Extractables and leachables may originate from process- and product-contact materials throughout the manufacturing of a biotechnology product. These include upstream processes through purification steps to final fill and finish of the final drug product. From left to right, the symbols represent vial for master cell bank, bioreactor, filter housing, chromatography columns, ultrafiltration/diafiltration skid, carboys for drug substance storage, and primary container for drug product.
Direct and Indirect E&L Risk to Patient Safety
From toxicology perspectives, the inherent toxicity of the organic compounds, if present at sufficient high levels in a final drug product, may pose direct risk to patient safety. On the other hand, the risk of elemental impurities, including metals, is considered low at the levels reported for biotechnologically derived products (7, 31). The obvious reasons are (1) biotechnology products do not typically use metal catalysts, and (2) inorganic compounds may be used upstream in cell culture media, but the elemental impurities are cleared in downstream chromatography steps and ultrafiltration/diafiltration (UF/DF) steps. In addition, in-process elemental impurities arising from buffer solutions in typical platform processes have been shown to be effectively cleared (>99% removal) post-UF/DF. Finally, drug products on stability are monitored for elemental impurities. The stability data typically show elemental impurities below the low limits of quantitation (LOQ) by ICP-MS. When detected, the levels are not of toxicological concern when reviewed against the guidelines by the ICH (7) or Dietary Reference Intakes (DRI) (21) by the IOM. A less obvious reason is that these elemental impurities may pose an indirect risk as a result of structural modification of the therapeutic protein that may in turn lead to loss of efficacy and/or increased immunogenicity in the patient population at risk. Such indirect risk is not readily apparent but can be evaluated by product impact assessments against the relevant critical quality attributes of the therapeutic protein. In addition, assessments by a clinical immunology function also play a critical role not only during clinical development but also in post-marketing surveillance.
The Orthogonal Approach for E&L Data Analysis
Primary containers or drug delivery devices such as prefilled syringes are composed of multiple types of materials. In a prefilled syringe, a drug product is exposed directly to glass or plastic barrel, silicone oil lubrication, elastomeric stopper, and stainless steel needle, and indirectly to the needle shield and residues from tools used to manufacture or process the syringe (Figure 3). Information regarding the raw materials used to manufacture individual components is often incomplete.

Components of a prefilled syringe system require a wide array of analytical techniques to enable the orthogonal approach for analysis of extractables and leachables.
Extractable substances from the direct or indirect contact materials can have varying properties that require an orthogonal analytical approach for identification. A single analytical technique is not capable of identifying all the extracted substances. Instead, an array of techniques and methodologies (Table I) is leveraged to identify and obtain an extractable profile of a container system. Typically, elements (including metals) associated with inorganic materials are determined by ICP-MS, organic substances are identified with LC-MS, GC-MS, and/or HPLC, and polymers can be detected by size exclusion/gel permeation chromatography and/or mass spectrometry. Detectors such as ELSD (32) or total organic carbon (TOC) (33) can be used to estimate total amount of extracted materials. Many enhancements such as detectors, ionization, probes, modes, and sample preparation can be used to extend the analytical capabilities of the techniques listed in Table I.
The Advantages of Studying Assembled and Processed Systems
From a wide variety of materials that were tested per USP and ISO and met such specifications, a database of organic compounds from vendor validation guides and from internal extraction studies was compiled. The database was focused on organic compounds reported as volatile, semi-volatile, and non-volatile based on the wide array of analytical techniques. The vendors and internal extraction studies identified and quantified over 200 organic extractable compounds. The extraction studies were typically conducted with fully assembled and processed systems, and these include prefilled syringe systems, vial stopper systems, drug delivery devices, and single-use bioprocess bags. The typical processing steps such as gamma irradiation or autoclaving would have occurred prior to the extraction studies. For prefilled syringes, the full system would consist of the syringe barrel, needle and shield, and plunger stopper. For a drug delivery device, the test system would only include the fluid-contact components. To the extent possible, a single-use bioprocess system would include the bag, tubing, and connectors. The key advantage in studying an assembled system is to expose only the product-contact surfaces to the extraction solvents. This approach is very effective in reducing the number of extractable compounds to only those that are likely to migrate and interact with the drug product. Using this approach, extraction profiles were generated for representative systems as a basis for toxicology assessments.
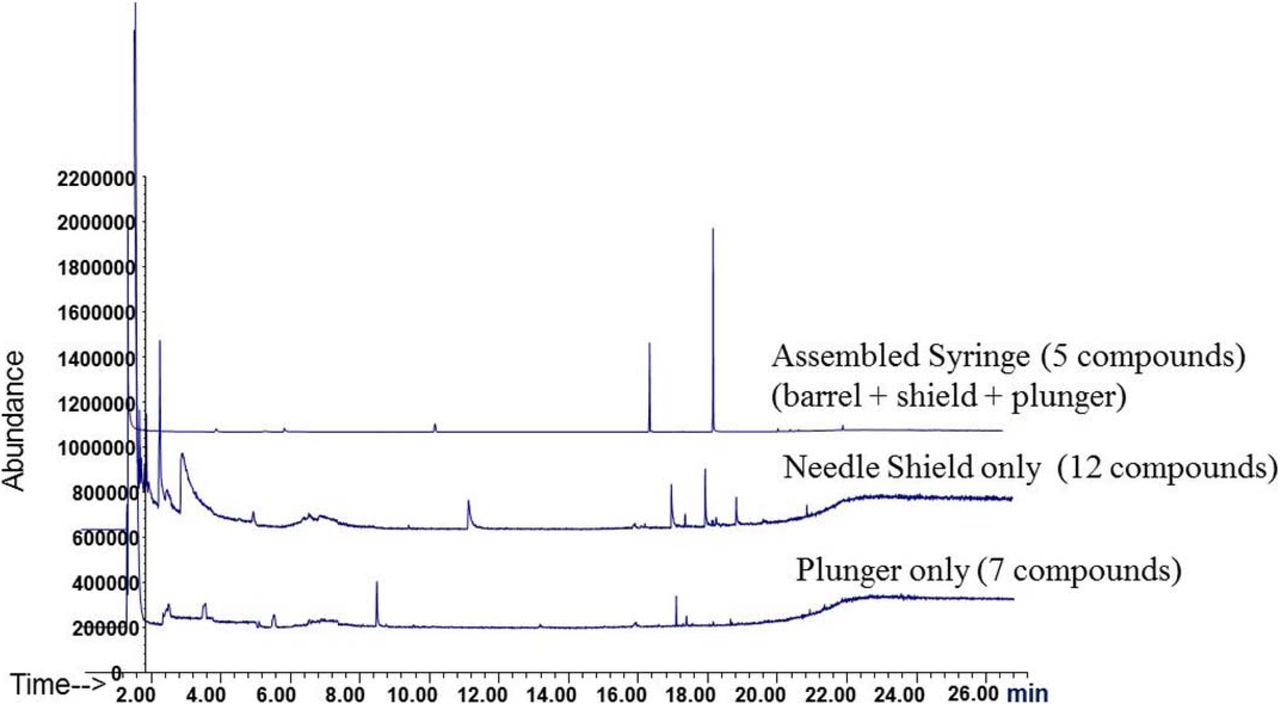
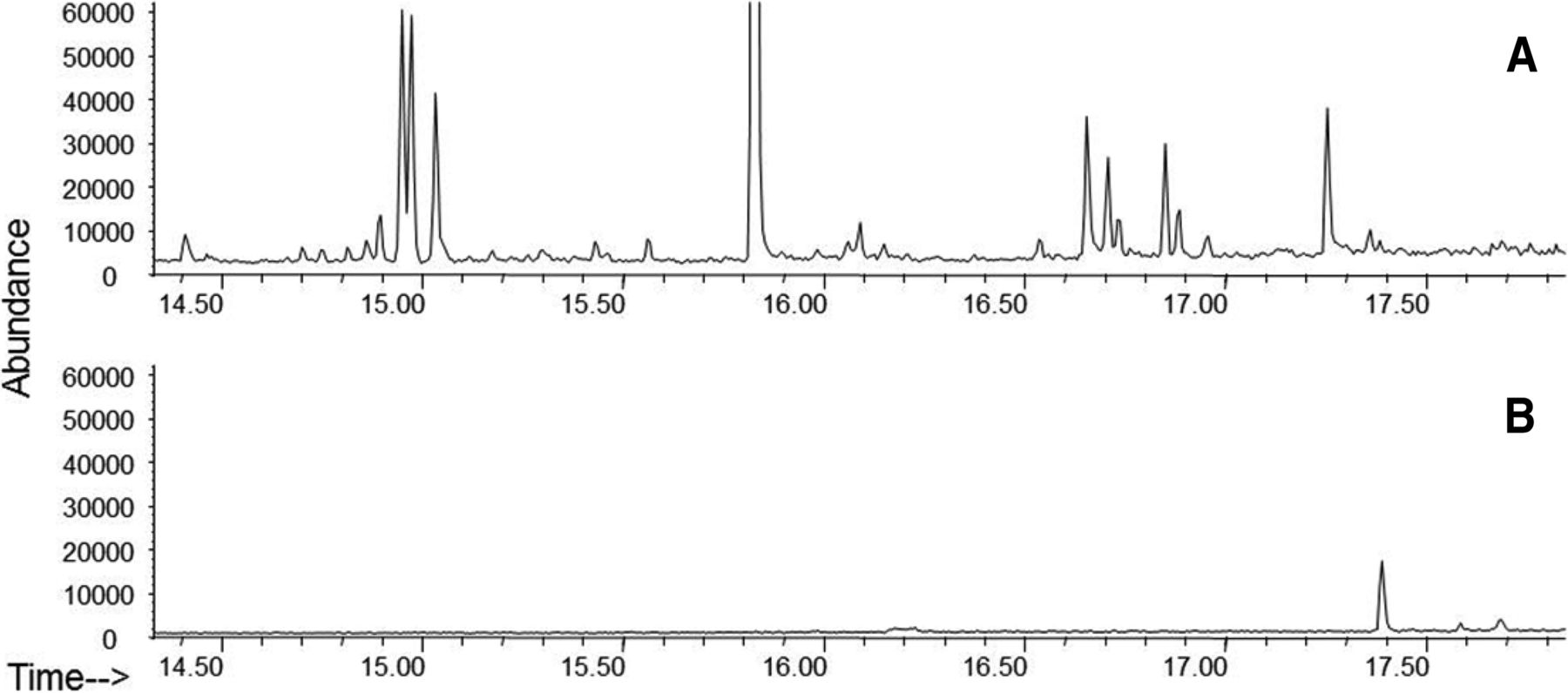
In a case study using a prefilled syringe system (Figure 4), comparing the extraction profile of the assembled system with those of the individual components enables the identification of the extractables and to link to their possible source(s). Individually, syringe components yielded a total of 24 compounds while the assembled system produced only five compounds. It was easily deduced that the five compounds originated from the syringe barrel because they were not present in the needle shield or stopper/plunger. Similar extractable study comparisons can be further pursued to isolate the exact extractable source on the barrel (i.e., adhesive, lubricant, needle, residuals from tools or manufacturing process). Identification of source provides understanding and troubleshooting of the materials and ultimately improves container process development.

GC-MS chromatograms of extracts from an assembled syringe system, individual needle shield, and individual plunger. Analysis of assembled systems may contain fewer extracted substances relative to the sum of the individual components. The use of assembled systems may be more relevant in predicting leachables. In addition, the source of extracted substances can be determined. All extractions were performed using identical extraction solvent, time, and temperature.
Extractables Studies
Selecting an appropriate extraction time and temperature is needed to accelerate aging of the container and generate extractables or, ideally, representative leachables. The Arrhenius rate equation is commonly accepted for supporting shelf life and expiration dating of sterile barrier systems for medical devices. The Arrhenius rate equation states that every 10 °C increase in temperature doubles the rate of a chemical reaction, and this calculation is leveraged to approximate an appropriate extraction temperature and duration for simulating an accelerated contact exposure, migration, reactivity, and aging process.
Biopharmaceutical products are aqueous-based formulations with typical shelf lives of 1–5 years at recommended storage temperature of 5 ± 3 °C. Applying the Arrhenius equation, an accelerated temperature of 70 °C for 4 days mimics 1 year storage at 5 °C. However, extending incubation time beyond 4 days and/or raising the temperature above 70 °C may result in degrading product-contact components and/or compromise the closure integrity. Degradation profiles under such harsh conditions would not be meaningful for predicting leachables. In contrast, aqueous–organic mixtures designed to mimic formulation conditions generate significant quantities of extractables pertinent to the understanding of the contact surfaces and components (34).
Aqueous mixtures of organic solvents such as acetonitrile (ACN) or ethanol (EtOH) are more efficient than pure water in extracting organic compounds such as butylated hydroxytoluene (BHT) from elastomeric plungers (Figure 5). Increasing the organic content in the extraction solution led to increased amount of extracted BHT. Extraction at 70 °C for 4 days resulted in an exponentially higher amount of BHT with the 40% organic solution compared with pure water. The use of a 40% organic solution is capable of extracting relatively large quantities of individual extractables, which is advantageous for identification. Amounts extracted are expected to be higher than those in the leachable studies. Therefore, extraction data by experimental design are conservative for toxicology and product impact assessments.

Amount of BHT extracted from assembled syringe systems using aqueous extraction solutions containing a total of 0-40% organic solvents at 70 °C for 4 days.
The use of a 40% organic solution has advantages over pure isopropanol that is a recommended solvent used in USP <381> testing. Extraction of elastomer stoppers was compared for USP <381> conditions (100% isopropanol solvent refluxing at 80.4 °C) and 40% organic at 70 °C for 4 days by GC-MS analyses (Figure 6). As expected, the USP method extracted significantly larger numbers and amounts of volatile extractables. In contrast, only one compound was extracted with 40% organic solution at 70 °C for 4 days. The USP <381> extraction condition can lead to an array of irrelevant extractables that are not predictive of realistic leachables. The identification and assessment of the irrelevant extractables provide little value in the prediction of leachables.

GC/MS chromatograms of extracts from elastomer plungers using (a) USP <381> 100% isopropanol, reflux (80.4 °C) for 30 min and (b) acetonitrile/ethanol (20%/20%), 4 days at 70 °C.
Extraction using 100% organic solvents, such as isopropanol, hexane, or ethanol, is not appropriate due to the sheer number of extracted materials that are unlikely to be leachable and would be inappropriate for mimicking biopharmaceutical formulations. Use of 100% water is compatible with inorganic material, but not a viable option for extracting organic compounds due to their low solubility in water. Although biopharmaceutical drug products are aqueous-based, they may contain protein and/or organic excipients that may act as surfactants and enhance the solubility of organic substances. Therefore, a blend of organic solvents is recommended for extraction of organic compounds. In addition, water is recommended for extraction of elemental impurities.
Leachables Studies
The appropriate design of the extraction studies with assembled systems described in the previous section has demonstrated good predictability for leachables in drug products, as illustrated with the case studies below.
Case Study 1.
Extractable and accelerated leachable profiles were generated from assembled glass syringes using 40% aqueous organic solutions at different pHs (70 °C for 4 days) and four biopharmaceutical drug products (37 °C for 90 days). The results are shown in Table II. The extracted/leached compounds were identical, with similar quantities for the 40% aqueous organic solutions compared with the drug products. In these cases, the extractables predicted the accelerated leachables from assembled syringe systems.
Comparison of the Extractables and Accelerated Leachables Results of a Prefilled Syringe System
Case Study 2.
A glass syringe system and novel drug delivery device (containing many polymer components) were evaluated for extractables and real-time leachables. Extractions were performed using solutions of 40% aqueous organic solutions at various pH levels at 70 °C for 4 days. Ten organic extractables were identified from the syringe. Analytical methods were developed to quantitate each substance in the corresponding drug formulations based on accuracy, precision, spike-recovery, carryover, limits of detection (LOD), LOQ, specificity, and linearity. Drug products were stored at 5 ± 3 °C up to 3 years or shelf life. None of the 10 organic extractables were detected (LOD: 0.1 ppm) in drug product after 3 years. In addition, general screening of drug products did not detect any new leachables. Toxicology assessment indicated that only one of the 10 reported compounds was of toxicological concern, namely, target organ toxicity, genetic toxicity, reproductive/developmental toxicity, and/or immunotoxicity. This compound would have served as a good indicator for the real-time study.
Case Study 3.
In a similar study on a novel drug delivery device, 61 compounds were extracted. A toxicology review on all the compounds identified only four compounds of concern. The reasonable and pragmatic alternative was to develop methods specific for the four compounds in a drug product. Using this risk-based approach, all four compounds were <LOD in the drug product.
Case Study 4.
While the leachables were generally a small subset of the extractables, it is important to understand the likely origins of the leachables. Adhesives are used in syringes to glue the metallic needle to the glass syringe. In this case study, Irgacure 184 was used for UV-curing of the adhesives. Irgacure 184 is a photo initiator commonly used for this purpose (35). All the extracted organic compounds were related to the adhesive process and/or Irgacure 184 (see Figure 7). As a result of this observation, the syringe supplier optimized the UV adhesive curing process to minimize these leachables.

Identified extractables/leachables and relationship to the source material. Irgacure 184 is commonly used as a photo-initiator for UV-curing adhesives (it induces polymerization), which is used to glue the metallic needle to the glass barrel.
Decision Tree for Toxicology Assessment
Depending on the complexity of the components/systems, as many as 61 organic compounds were detected per system. Therefore, it was absolutely critical that the toxicology reviews addressed the compounds of concern commensurate with the levels that might translate to patient risk. Figure 1 shows a decision tree for a typical toxicology review on organic extractable compounds. The decision tree is built on the best practices for assessing cancer and non-cancer risks for large datasets of organic compounds including pesticides, foods, drugs, food additives, and industrial and environmental chemicals (15⇓⇓⇓–19). The series of questions in the decision tree helps to effectively target the compounds with significant toxicity taking into account the levels of toxicological concern. For data-rich compounds, literature data were the primary source of the review. In instances where data gaps were identified, particularly genotoxicity, in silico analysis with Toxtree (13) was used for structural alerts. Toxtree is an acceptable screening tool for quantitative structure–activity relationship [(Q)SAR] analysis under ICH M7 (36, 37). Expert knowledge and an additional (Q)SAR program per ICH M7 are used if Toxtree predicts a structural alert.
In the resulting dataset of over 200 E&L compounds, most were estimated below 1.5 μg/system, the lowest (most conservative) safety concern threshold (SCT) of 1.5 μg/system for genotoxicants for PODP (10). In the cases of the prefilled syringes and vial stopper systems, this estimate often calculated to patient exposure of less than 1.5 μg/day, assuming daily, chronic administration of the drug product in these systems for a lifetime. This SCT for genotoxic impurities was the first level to screen out unstudied compounds at levels of no toxicological significance. Then literature reviews were conducted on the compounds with levels estimated at greater than the SCT of 1.5 μg/system.
Permitted Daily Exposure (PDE) and Cramer Classifications
The majority of the identified compounds (151 out of 220) in the dataset had adequate information for risk assessment purposes. Many of these compounds had acceptable uses as polymer additives and as direct and/or indirect food additives. For materials that had been sterilized or gamma-irradiated, their profiles often showed degradation products of the additives. The commonly observed compounds included caprolactam, fatty acid amides, polyethylene glycol (PEG)-containing substances (mw < 600), poly(1,4-butanediol) fragments, PDMSs (linear and cyclic), methacrylates, and degradation products of antioxidants (BHT, Irgafos 168, Irganox 1010, and Irganox 1076). However, the structures of many of the degradation products could only be tentatively identified. Given that only trace levels exist, even under harsh extraction conditions, the qualitative and semi-quantitative profiles were adequate for risk assessment purposes.
Upon a full literature review and evidence of no mutagenic potential, pPDE values were derived following the principles and methods described in ICH Q3C(R5) (5). In addition to the modifying factors to account for interspecies extrapolation (F1), individual variability (F2), study duration (F3), severe toxicity (F4), no-effect level (F5), an additional factor (F6) was used to account for bioavailability. In the derivation of pPDE, it was critical to use available literature information on the absorption, distribution, metabolism, and elimination (ADME) of the compound. If no ADME information was available, a default value of 10 was used for F6. Therefore, the pPDE was calculated as follows:
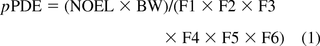 where pPDE is the parenteral PDE in μg/day
where pPDE is the parenteral PDE in μg/day
NOEL is no-effect level in mg/kg body weight/day
BW is 60 kg body weight.
The use of 60 kg as the body weight is based on FDA guidance: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers (38). As a result, the dosing regimen of marketed products is often based on 60 kg based on the clinical trials. Using this approach, pPDE values were derived for 103 compounds with adequate toxicity data (out of 151 in the dataset). The derived pPDE for the dataset (103 out of 151) ranged from 3 μg/day to over 1.2 g/day. A few compounds also existed as isomers or related structures, namely, cresol isomers, ricinoleic acid isomers, octene isomers, X-tert-butylcyclohexanol isomers, heptadecasphinagine isomers, and compounds related to 4,4′-thiobis(6-tert-butyl-m-cresol). The literature on these compounds did not reveal significant differences in the toxicity profiles of the isomeric or related structures. Therefore, single pPDE values were derived. Appendix 1 lists the 103 compounds, in ascending CAS numbers, the derived pPDE values,and the modifying factors used in the derivation. Appendix 2 lists 45 compounds (out of 151 in the dataset) for which insufficient information was available, and therefore we used in silico predictions for Cramer classifications using Toxtree (13). It is important to note that the compounds in both Appendices 1 and 2 were not considered to be DNA-reactive (mutagenic) impurities by ICH M7 guideline (6) either by literature review or by in silico predictions for structural alerts.
Based on the derived pPDE, the compounds were then classified into Cramer classes.15–19
The Cramer Thresholds of Toxicological Concern (TTC) are as follows:
Cramer Class I (low toxicity): 1800 μg/day
Cramer Class II (intermediate toxicity): 540 μg/day
Cramer Class III (high toxicity): 90 μg/day.
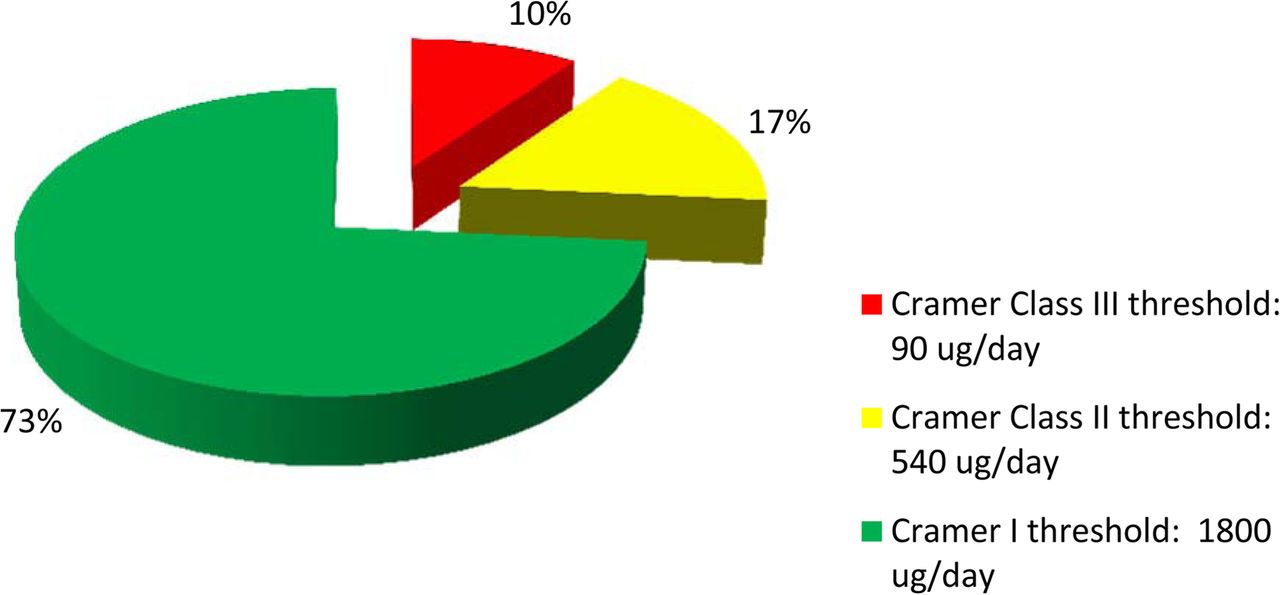
Figure 8 shows the risk analysis of the dataset (n = 151) by Cramer classes. The analysis showed that only about 10% of the identified compounds were in Cramer Class III. The majority (73%) was in Cramer Class I, and relatively low percentage (17%) was in Cramer Class II. Based on the risk analysis, it became apparent that high-concern compounds were only a small subset of the dataset. Such compounds may pose risk to patient safety if they leached into a final drug product at levels exceeding the pPDE. In contrast to a recent publication by Jenke and Carlson (39) on extractables associated with materials used in pharmaceutical packaging, delivery, administration, and manufacturing systems, the dataset presented in this paper is based on materials certified for pharmaceutical and/or food contact applications. Such certification and compliance with current USP and/or ISO requirements help to eliminate many plastic additives of safety concerns. In addition, our extraction conditions and methodologies are robust and relevant for biologics. Therefore, the lower distribution of Cramer Class III compounds in this dataset is not totally unexpected.

The risk analysis of the organic extractables (n = 151) from process- and product-contact materials showed that the majority of the compounds was of Cramer Class I (low toxicity), while only a low percentage was of Cramer Class III (high toxicity).
Compounds in Cramer Class III are characterized by reactive functional groups that can lead to significant toxicity (18). The underlying concept is that the interactions of a reactive chemical with critical cellular targets is the molecular initiating event leading to toxicity (27). The most critical and prevalent targets are macromolecules, especially DNA and proteins (40). The best known reactive functional groups include aliphatic secondary amino-, cyano-, N-nitroso-, diazo-, triazeno-, quarternary N, strain-ringed lactones, epoxides, quinones, and α,β-unsaturated ketones (18, 40). Many of these represent structural alerts for Michael acceptors (27) or electrophiles capable of covalent protein binding (25, 26).
In the development of the Cramer classification, Class III was also designed to capture structures with propensity for metabolic bioactivation to potentially toxic chemical entities (18). The expert working group examined many factors, such as metabolism and accumulation, structural alerts, and toxicity endpoints, and concluded that specific considerations of metabolism and accumulation were not necessary in the application of the Cramer TTC (18). In this regard, the oral Crammer TTC would apply to the pPDE as derived for the leachables.
ICH Q3C defines PDE (not route-specific) as an acceptable intake of an impurity (e.g., residual solvent) in a pharmaceutical product. Because solvents gain rapid distribution and systemic absorption, ICH Q3C does not need to address the different routes of administration. On the other hand, leachables may be volatile, semi-volatile, or non-volatile organic compounds with varying ADME profiles. Therefore, we use F6 to adjust for bioavailability. pPDE is derived for exposure on a daily basis for a lifetime. Therefore it is an extremely conservative limit for impurities in biotechnology products, which are used only on intermittent basis. In addition, the actual processes in the manufacturing, transportation, and storage of the biotechnology products are carried out under much milder conditions relative to the extraction study conditions.
Transformed or Degradation Products
Frequently an extractable profile showed clusters of structural analogs, for example, transformed products of BHT) and Irgafos 168. If there were adequate literature data on the analog, an individual pPDE would be derived. Otherwise, the parent compound would serve as the surrogate or indicator compound for the cluster. In such cases, the total mass estimate of the cluster would be assessed against the pPDE derived for the surrogate.
Concept of Action Threshold in Reducing E&L Risk to Product Quality
The analysis of the dataset on the identified extractables with pPDE and Cramer classifications has led to the concept of action threshold to target and monitor leachables in drug products on stability until expiry. It is important to emphasize that the proposed concept of action threshold applies to only organic leachables. Elemental impurities (and transition metals) are discussed under Product Impact Assessment.
Using the risk-based approach, a toxicology assessment serves two key purposes: (1) screen components/systems for hazard identification and qualification purposes, and (2) identify compounds of potential toxicological concern for leachable studies for drug products on stability. However, as discussed previously, the non-genotoxic leachables have a wide range of pPDE (≤1.5 μg/day to >1 g/day). Therefore, it becomes apparent that while pPDE values serve well in the qualification of individual leachables as impurities from a product safety perspective, they cannot serve as general thresholds from product quality perspective. This is particularly true for biotechnology products that are susceptible to structural modification by very low levels of impurities.
Leachables that pose greatest risk of structural modifications are those that may form covalent bonds with proteins. These are discussed under the Product Impact Assessment section. Specifically, organic compounds that are capable of forming covalent bonds with proteins are electrophiles or Michael acceptors (26, 27). Experience with a wide variety of components and systems show that these are a very small subset of the organic extractables. Such examples include acrylic acid, methacrylic acid, 1,6-hexanediol diacrylate, and dibutylmaleate (Figure 9).

Electrophiles are a small subset of the reported extractables and leachables. Examples shown here were based on product impact assessment of extractables profiles of assembled systems.
In order to assess the impact to both product safety and quality, it is proposed to use the action threshold in the leachables testing of final drug products on stability to target the high-risk compounds:
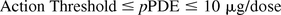 The unit for action threshold is microgram per dose (μg/dose), assuming a daily dose for a lifetime. Because even high-risk compounds (Cramer Class III) may have thresholds as high as 90 μg/day, it is proposed that action thresholds not exceed 10 μg/dose, about a factor of 10 below the Cramer Class III threshold. In the foregoing discussion, interaction with the critical cellular proteins is discussed as the molecular initiating event for Cramer Class III compounds. By applying a factor of 10 to the Cramer Class III TTC, the action threshold reduces the risk of initiating the molecular event. This limit for action threshold also applies to the relatively safe leachables (high pPDE) in order to mitigate their risk to product quality if present at high levels. With this risk-based approach, action thresholds not only protect patient safety but also serve to trigger quality investigations that may examine structural modifications, critical quality attributes, and risk for immunogenicity.
The unit for action threshold is microgram per dose (μg/dose), assuming a daily dose for a lifetime. Because even high-risk compounds (Cramer Class III) may have thresholds as high as 90 μg/day, it is proposed that action thresholds not exceed 10 μg/dose, about a factor of 10 below the Cramer Class III threshold. In the foregoing discussion, interaction with the critical cellular proteins is discussed as the molecular initiating event for Cramer Class III compounds. By applying a factor of 10 to the Cramer Class III TTC, the action threshold reduces the risk of initiating the molecular event. This limit for action threshold also applies to the relatively safe leachables (high pPDE) in order to mitigate their risk to product quality if present at high levels. With this risk-based approach, action thresholds not only protect patient safety but also serve to trigger quality investigations that may examine structural modifications, critical quality attributes, and risk for immunogenicity.
It is important to note that not all extractable profiles will have compounds in Cramer Class III. In such cases, compounds of lower concern (e.g., Cramer Class II) may be selected as potential leachables in stability studies. Also, the most abundant, but innocuous, compounds (e.g., Cramer Class I) may be considered in targeting leachables (41).
Product Impact Assessment—General Considerations
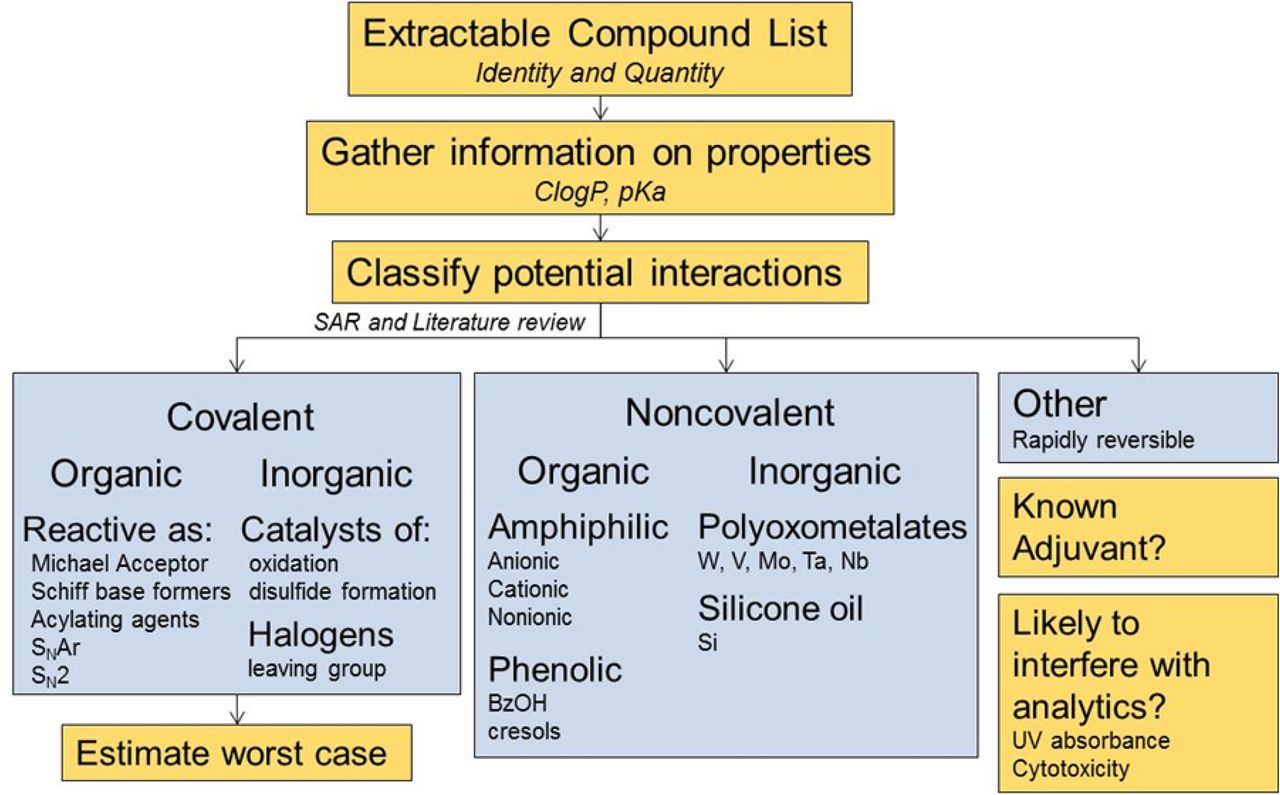
The primary goal of a product impact assessment is to evaluate the potential of the extractables to interact with a therapeutic protein. Conclusions of these assessments are intended to support the determination of any need for either further characterization of the proposed mechanisms of interaction, or monitoring of adverse impact of leachables on the drug product on a routine basis. The clinical risks associated with a loss of efficacy, which requires specific knowledge of key interactions governing pharmacodynamics and pharmacokintecs of the therapeutic molecule, or increased immunogenicity of a structurally modified protein, are difficult to assess from the chemical interactions alone. However, critical product attributes may be monitored for clinical relevance. A schematic representation of the assessment strategy is presented in Figure 10. It requires knowledge of both the extractables and the product attributes. Because of the susceptibility of biotechnology products to chemical interactions leading to structural modifications, it is the purpose of a product impact assessment to classify the potential interactions and assess the level of concern for protein modification. In this context, there are two major types of protein interactions: covalent binding and noncovalent binding. Both organic compounds and elemental impurities may participate in covalent and/or noncovalent binding. Covalent interactions are irreversible, stabilized by a single point of contact. Noncovalent interactions are generally reversible and typically require multiple weak interactions involving electrostatic, hydrophobic, and/or hydrogen bonds to become stable. Because of the irreversible nature of the protein modification, covalent binding presents a higher risk of affecting product quality attributes as compared to noncovalent binding.

Schematic representation of the proposed strategy for assessing the potential impact of extractable compounds on product attributes. Once the identity and an estimate of the quantity for individual extractable compounds and elemental impuritiesare available, interactions with protein therapeutics (and formulation components, if appropriate) can be categorized as covalent, noncovalent, or other (rapidly reversible and likely of low concern). Worst-case scenarios can be estimated based on molar ratios of extracted components and mechanisms of interaction with the drug product. Each compound or element should also be considered for potential adjuvant effects or possible interference with analytics used for routine monitoring.
From the extraction studies, the profiles of the organic compounds and elemental impurities are screened for potential protein interactions. The dataset was comprised of 216 compounds, many with only tentative chemical structures and no other chemical identification. Based on the schematic presented in Figure 10, the majority of compounds were characterized as nonreactive (n = 169). The remaining compounds were characterized as reactive based on potential to form covalent bonds with proteins. Six of the reactive compounds may be able to interact by more than one mechanism. The reactive compounds were characterized as Michael acceptors (n = 22), Schiff base formers (n = 12), acylating agents (n = 7), agents that interact through aliphatic nucleophilic substitution (n = 5), agents that interact through aromatic nucleophilic substitution (n = 1), and transition metals (n = 8). The reactive compounds are listed in Table III, and the six compounds with multiple mechanisms are also listed under the individual mechanisms that apply (Table III). The mechanisms of interaction for these different groups are discussed in more detail in the following sections.
List of Compounds That Can Potentially Form Covalent Modification of Proteins
If the interaction is covalent, the extent of reaction is estimated with the worst-case assumption that the entire mass estimate of the compound became covalently bound to the protein. If the molar ratio of the potentially reactive compound(s) exceeds 2% (mol:mol), considerations would be given to further characterization studies including mechanistic studies and/or accelerated stability testing. The 2% (mol:mol) threshold for further experimentation is based on the assumption that a non-targeted peptide map monitored by UV-absorbance could detect this level of modification at a single residue. By using targeted LC-MS, the sensitivity of peptide map techniques can be reduced to 0.1–0.001% of modified peptide, in other words, levels consistent with naturally occurring amino acid mis-incorporations observed in recombinant proteins expressed in Escherichia coli, monoclonal antibodies (mAbs) expressed in mammalian cells, and human serum albumin purified from human subjects (42).
Most protein therapeutics are confined to a relatively narrow range of solution conformations on the basis of formulation development and controlled storage conditions chosen to maximize stability, and usually solubility, of the molecule. While noncovalent interactions between the extractable components of the primary container and the protein therapeutic or formulation excipients are generally expected to be reversible, gross changes in protein structure can be detected by either size exclusion chromatography (SEC) or particle inspection. These methods are usually a major part of typical programs to monitor stability of the active ingredient. Additional characterization of potential interactions between protein and extractables may be indicated in extreme cases.
While there are assays to easily detect changes in the pharmacological properties of a chemically modified protein, assessment for immunogenicity is more difficult. The approach described here provides a scientific basis to predict the possibility of protein modification, which may be a key molecular event leading to increased immunogenicity of certain proteins. There is, as of yet, no clear threshold of concern for hapten-mediated breakage of self-tolerance for either humans or animals. However, whether the protein modification leads to increased immunogenicity or reduced efficacy must be evaluated by product attributes relevant to such clinical risks.
Covalent Interactions—General Mechanisms
Most, if not all, nonenzymatic covalent reactions between proteins and organic compounds are nucleophilic substitutions in which the functional groups of amino acid side chains serve as the nucleophile and the chemical modifier is the electrophile (43). The N- and C- terminus, and functional side chains of arginine, lysine, histidine, cysteine, tyrosine, glutamic acid, and aspartic acid, are the most chemically active.
Protonation decreases the nucleophilicity of a species so the pH of the medium will affect both the rate of reaction and the probability that the reaction will occur. As shown in Table IV, the functional groups with relatively high pKas (the phenolic hydroxyl of tyrosine, the ϵ-amino of lysine, and the guanido group of arginine) will be fully protonated under pH conditions typically used for formulation of protein therapeutics and would react slowly with low concentrations of electrophilic compounds. Carboxylic acids on the C-terminus, glutamic acid, and some aspartic acids will be deprotonated but are generally considered to be relatively “weak” nucleophiles. (Oxygen is relatively electronegative, and the electron density is distributed by resonance throughout the carboxyl system and therefore less likely to share unbound electrons in a displacement reaction.) Reaction rates at the amino group on the N-terminus, the sulfhydryl group of cysteine, and the imidazolyl group of histidine would be expected to exhibit variable reactivity over the pH ranges typically used in formulation of protein therapeutics.
Percent of Deprotonated Amino Acid Side Chains over the pH Range Typically Used for Formulation of Protein Therapeutics (Calculated from the Henderson-Hasselbach Equation According to Skoog and Wichman, 1986)
Directed experiments to evaluate the kinetics of a potential reaction can provide additional insight into the relative amount of risk associated with the presence of a specific compound. Liu et al. (44) identified acrylic acid, a known Michael acceptor, as one compound leaching from an acrylic adhesive commonly used to attach the needle of prefilled syringes. Incubation of a model IgG2 antibody, 15 mg/mL, at 45 °C for 45 days in a pH 5 acetate buffer with 5 μg/mL (0.7 mol per mol mAb) or 250 μg/mL (35 mol per mol mAb) acrylic acid followed by peptide mapping using LC-MS identified several sites of covalent modification distributed throughout the molecule including two residues in the complementarity defining region and two residues in the FcRn binding site (H316 and H441).
Modifications observed at 0.7 mol acrylic acid per mol antibody (70% mol:mol) did not exceed 0.3% mol:mol of total modified antibody. This suggests that a 2% mol:mol threshold of investigation may in fact be quite conservative with respect to pharmacokintetic and pharmacodynamic properties of the molecule but should be confirmed by routine monitoring in a stability program to provide additional assurances that chemical modifications are not focused in specific areas crucial to the biological function.
Bromide and chloride are occasionally observed by ICP-MS in aqueous extracts from primary containers or devices containing halobutyl elastomer components and may not be clearly associated with any of the organic compounds observed in the extractable profile. Because halogens (Cl, Br, and I) are a good leaving group in nucleophilic substitutions, a product impact assessment would disregard the nature of the halogen (i.e., organic or elemental) but use a worst-case scenario using the molar ratio of halogen and drug product. As noted above, a 2% (mol:mol) ratio of halogen to active ingredient suggests that further characterization or monitoring may be indicated.
Redox active transition metals identified in aqueous extracts of a primary container can contribute to the oxidation of protein therapeutics. Chain-initiating radicals can be generated through exposure of the system to redox-active transition metals (45, 46). Once an initiator radical has formed many compounds, including the protein therapeutic itself, it can participate in the propagation and termination phases of the chain oxidation process. The levels of these metals need not be very high.
Redox-active transition metals can also participate in site-directed oxidation of neighboring amino acids by binding to Gly, Asp, His, or Cys residues (46, 47). Although dependent on additional factors, metal-catalyzed site-directed oxidations can apparently take place in the presence of <30 ppb transition metals (48).
Noncovalent Interactions—General Mechanisms
Because of the transient and reversible nature of nonconvalent interactions, the risk to product impact is considered low relative to covalent interactions. The solution dynamics of any given protein are complex, with the existence of multiple equilibrium states largely dependent on the balance of local surface charges (either attractive or repulsive) and the attractive interactions attributable to hydrophobic amino acid residues.
Organic Extractables—Considerations for Noncovalent Interactions
Organic compounds identified in the extraction studies can be classified based on their amphipathic properties. Amphiphiles can be further divided into anionic, cationic, nonionic, or phenolic amphiphiles for the purpose of evaluating their potential to interact with protein therapeutics. Compounds that do not meet these criteria can be considered to have no significant interaction with the product.
Anionic amphiphiles include compounds with an anionic (carboxylic acid, phosphate, or sulfate, for example) group at one end of the molecule and a hydrophobic group (defined as six uninterrupted carbons with no oxygen or nitrogen components attached) at the other end. The ionic character of these compounds in the system will be determined by the pKa of the polar group and the pH of the formulation. Binding of monomers of anionic amphiphiles to protein would involve electrostatic interaction with cationic amino acid side chains (Lys, Arg, His) and nearby hydrophobic patches on the protein (including the methylene backbone of the amino acid itself). Using surfactant protein interactions as a model (49), detectable changes in structure would not be expected until 4–10 monomers were bound to the protein. In either charged or neutral state these compounds might be expected to preferentially partition into the air:water interface along with any neutral surfactant (e.g., polysorbate) in the formulation, or into mixed micelles.
Fatty acids, which are often used as slip agents in the manufacture of polymers films and devices with moving parts, represent anionic amphiphiles that require special consideration. Kishore et al. (50) have shown that fatty acids present in a molar ratio >0.1:1 relative to polysorbate 20 can lead to the formation of insoluble protein aggregates of mAbs formulated at pH 6.
Cationic amphiphiles include compounds with a cationic group (most likely primary amine) at one end of the molecule and a hydrophobic group (defined as six uninterrupted carbons with no oxygen or nitrogen components attached) at the other end. The ionic character of these compounds in the system will be determined by the pKa of the polar group and the pH of the formulation, and electrostatic interactions with the protein will be directed at anionic amino acids (Asp, Glu, and any post-translational modifications that introduce an anionic group to the protein). As discussed for anionic amphiphiles, nearby hydrophobic patches on the protein would be expected to stabilize the interaction but detectable changes in protein structure would not be expected until 4–10 monomers were bound. In either charged or neutral state, cationic amphiphiles might also be expected to preferentially partition into the air:water interface along with any neutral surfactant (e.g., polysorbate) in the formulation, or into mixed micelles with the surfactant.
Nonionic amphiphiles, like nonionic surfactants, will bind to therapeutic proteins primarily via hydrophobic interactions. The interactions of nonionic amphiphiles and proteins are expected to be weaker than those of ionic amphiphiles and require higher protein-to-amphiphile ratios to effect a change in protein structure. Nonionic amphiphiles would also be expected to preferentially partition into the air:water interface along with any neutral surfactant (e.g., polysorbate) in the formulation, or into mixed micelles with the surfactant.
Phenolic compounds such as the antibacterial preservatives benzyl alcohol and m-cresol can induce protein aggregation at relatively low concentrations by binding to the protein and favoring an increase in the level of partially unfolded, potentially aggregation-competent species rather than a global unfolding of protein (51). Typical antimicrobial concentrations are 0.9% benzyl alcohol (∼87 mM), 0.3% m-cresol (∼29 mM), and 0.5% phenol (∼57 mM) (52) Aromatic compounds with ionizable substitutions (e.g., carboxylic acids or amides) may form more stable complexes with proteins due to possible electrostatic interactions as well as hydrophobic interactions.
Elemental Impurities—Considerations for Noncovalent Interactions
Polyoxometalates are polyatomic ions, usually anions that consist of three or more transition metaloxyanions linked together by shared oxygen atoms to form a larger framework. The metal atoms are usually group 5 or group 6 transition metals in their high oxidation states. Examples include vanadium, niobium, tantalum, molybdenum, and tungsten. Tungsten has been associated with aggregation of a therapeutic protein in prefilled syringes (22). Aggregation appears to arise through electrostatic interaction between protein and the tungsten species and is dependent on pH of the buffer used, the tungsten species, tungsten concentration present, and the protein concentration. Increased aggregation was noticeable in the presence of 1 ppm tungsten extracted from a pin tool used to form the needle channel.
Spiking of epoetin alfa with sodium polytungstate or an extract of tungsten pins also induced formation of protein aggregates at concentration as low as ∼1 ppm (53).
Silicon is present in both the functional components (glass silicates, silicone components) and lubricants (silicone oil, PDMSs) of some primary containers and devices. Therefore, high levels of elemental silicon by ICP-MS above specification limits may warrant further characterization and/or stability studies.
Conclusion
We have studied the toxicological risk of extractable compounds from a wide array of primary container closure systems, on-body devices, bulk drug storage containers, and disposable single-use bioprocess materials that come into direct or indirect contact with drug substance or drug product. Based on the analysis of the pPDE values and the Cramer classification of over 150 compounds, we conclude that a toxicology threshold alone does not adequately protect quality attributes. Certain biotechnology products are susceptible to leachate-induced structural modifications, leading to impact on product quality and safety. The proposed concept on action threshold provides a science- and risk-based approach to balance the risk between product quality and safety during the development and throughout the lifecycle management of biotechnology products.
Conflict of Interest Declaration
The authors declare that they have no competing interests.
Appendix
Organic Compounds with Derived Parenteral Permitted Daily Exposure (pPDE)
Organic Compounds with Cramer Classifications by In Silico Predictions
Footnotes
FDA Disclaimer
Comments expressed in this review represent the opinions of the author and are not intended to endorse any product or to reflect FDA policy.
- © PDA, Inc. 2015
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.
- 55.
- 56.
- 57.
- 58.
- 59.
- 60.
- 61.
- 62.
- 63.
- 64.
- 65.
- 66.
- 67.
- 68.
- 69.
- 70.
- 71.
- 72.
- 73.
- 74.
- 75.
- 76.
- 77.
- 78.
- 79.
- 80.
- 81.
- 82.
- 83.
- 84.
- 85.
- 86.
- 87.
- 88.
- 89.
- 90.
- 91.
- 92.
- 93.
- 94.
- 95.
- 96.
- 97.
- 98.
- 99.
- 100.
- 101.
- 102.
- 103.
- 104.
- 105.
- 106.
- 107.
- 108.
- 109.
- 110.
- 111.
- 112.
- 113.
- 114.
- 115.
- 116.
- 117.
- 118.
- 119.
- 120.
- 121.
- 122.
- 123.
- 124.
- 125.
- 126.
- 127.
- 128.
- 129.
- 130.
- 131.
- 132.
- 133.
- 134.
- 135.
- 136.
- 137.
- 138.
- 139.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- In Silico Assessment of Biomolecule Reactivity with Leachables
- Development of Duration-Based Non-Mutagenic Thresholds of Toxicological Concern (TTCs) Relevant to Parenteral Extractables and Leachables (E&Ls)
- Holistic Extractables and Leachables Program: Evaluations of Prefilled Syringe Systems for Biotechnology Products
- Using Extractables Data of Sterile Filter Components for Scaling Calculations
- Extractables Screening of Polypropylene Resins Used in Pharmaceutical Packaging for Safety Hazards