
Abstract
Visible particles must be monitored as part of the control strategy for pharmaceutical products. Extraneous (foreign) particles are not acceptable in parenteral drug products. In biopharmaceuticals, formation of protein particles is recognized as an inherent quality attribute. All protein therapeutics contain particles that vary greatly in visibility and size from invisible (sub-micron) to visible (millimeter) and, as part of the control strategy, biopharmaceutical companies are required to monitor and minimize the presence of visible and sub-visible particles in their products. There is an industry-wide unmet need for particle standards for visual inspection of protein therapeutics. A new, semi-quantitative method using particle standards for assessing the levels of small, inherent visible particles is presented. This method can be used during product development to identify a formulation that minimizes particle formation and also during release and stability testing to monitor and control inherent proteinaceous visible particles.
LAY ABSTRACT: Visible particles must be monitored as part of the control strategy for parenteral biopharmaceutical drug products. In these products, formation of protein particles is a natural occurrence. All protein drugs contain particles that vary greatly in visibility and size from invisible (sub-micron) to visible (millimeter), and pharmaceutical companies are required to monitor and minimize the presence of visible and sub-visible particles in their products. There is an industry-wide unmet need for particle standards for visual inspection of protein drugs. A new, semi-quantitative method using particle standards for assessing the levels of small, naturally occurring visible particles is presented. This method can be used during drug development to identify a formulation that minimizes particle formation and also during testing of final clinical or commercial drug product to monitor and control naturally occurring proteinaceous visible particles.
- Proteins
- Protein aggregation
- Protein formulation
- Stability
- Biotechnology
- Visible particles
- Inherent visible particles
- Proteinaceous
- Particle standard
- Barium sulfate
Introduction
Control of visible particles is a major focus in the development of parenteral pharmaceuticals. Protein therapeutics are complicated by the fact that liquid protein products contain inherent particles of various sizes (1, 2). During protein therapeutics development, the amount of visible particles should be minimized throughout the shelf life of a product by development of an appropriate formulation.
According to the United States Pharmacopeia (USP) <1787> (3), visible particles can be broadly classified into three categories:
Inherent (existing as a permanent and inseparable element): Particles of the protein product inherent to the protein molecule that may be present on release or form over time
Intrinsic (inside, part of the whole): Impurities from the manufacturing process and/or container contact surfaces (for example, stainless steel, glass fragments, textile fibers, and rubber)
Extrinsic (outside, from the exterior): Foreign particles, not part of the process or product (for example, environmental contamination)
All three of these particle types need to be controlled. Intrinsic or extrinsic particles are not expected and should be removed during 100% inspection. Inherent particles are part of the product and may be expected. However, as illustrated in Figure 1, even the number and size of inherent particles should be controlled and the “snow globe” effect should be avoided. This paper presents a method that can be used to monitor and control the amount of small inherent particles.

Illustration of difference between inherent and extrinsic particles. Extrinsic particles should be eliminated as much as possible. Inherent particles are often expected in biological parenterals. However, the amount of inherent particles should be monitored and controlled and the “snow globe” affect should be avoided.
Current compendial guidance on visual inspection as it relates to inherent proteinaceous particles in biotherapeutics is not comprehensive, consistent, or globally accepted. The three major pharmacopoeias require that drug products are free from readily detectable foreign insoluble matter (1⇓⇓⇓⇓–6). The USP and Pharmacopoea Europaea (Ph. Eur.) allow for drug products to be “essentially” or “practically” free from visible particles. In an article written by authors from multiple pharmaceutical companies (7), it was argued that because there is limited evidence of patient risk, zero particles should be the goal when manufacturing injectable drug products rather than the requirement. USP <790> (4) defines essentially free as “no more than a specified number of units may be observed to contain visible particulates”, and it is generally related to the overall quality of the batch, not individual vials. Thus, these requirements generally do not pertain to the inherent, expected visible particles that are often found in protein therapeutics. The method presented here is intended for semi-quantitative determination of inherent proteinaceous particles rather than assessment of the quality of the batch with regards to intrinsic or extrinsic particles. This method is implemented after the 100% visual inspection and subsequent Acceptance Quality Limit (AQL) testing. It is used to define the limit on the amount of visible proteinaceous particles that can be present in a vial. In the case of proteinaceous particles, the presence of small visible particles tends to be consistent across the batch rather than present in a few vials. It is often impossible to count the visible particles because they are on the verge of visual acuity.
Regulatory authorities and industry recognize that proteins may form particles inherent to the product. Regulatory authorities expect manufacturers to implement a well-controlled procedure to ensure that “proteinaceous visible particles, when controlled within characterized limits (specifications), should not present a quality or safety concern” (1). Protein therapeutic product specifications may contain a qualitative description of visible particles such as “some or few proteinaceous particles may be present”. The method presented in this paper is an attempt to define the terms “some” or “few” to ensure robust monitoring of particles at release and over the shelf-life of the product.
The definition of “visible” particle varies among companies in the industry. USP <1787> (3) considers particles larger than 100 μm to be visible. However, the practical limit of visibility varies broadly depending on the test conditions and the nature of the visible particle (8). Particles smaller than 100 μm have been visualized by the naked eye under conditions of increased light intensity (9). A mechanism for achieving some degree of conformity across the industry is required, and one option is to create a set of particle standards for visual inspection of protein therapeutics. Some available particle standards are polystyrene latex beads, mono-disperse silica or polymethylmethacrylate beads, and polydisperse glass beads (3). All of these standards are uniform in size and shape (spheres), with a refractive index and shape that are distinctly different from particles formed by proteins (Figure 2) (10). MedImmune (Gaithersburg, MD) has developed particle standards (with similar visual properties to protein particles) that can be visually compared to protein therapeutics in clear glass vials for visual inspection. Barium sulfate was chosen as a result of evaluation of various materials. Proteinaceous particles significantly vary in morphology and size. The irregular shape of barium sulfate particles more closely resembles the shape of protein particles than monodisperse polystyrene beads (Figure 2). The refractive index of barium sulfate (1.6) is similar to the refractive index of protein (1.4) (11). Particles in barium sulfate standards are present as a heterogeneous population ranging from sub-micron to visible; they are inert and practically insoluble in water (12). As shown in Figure 5, barium sulfate particles tend to be smaller than 100 microns, similar to small, inherent proteinaceous particles present in parenteral biopharmaceuticals.

The irregular and elongated shape of barium sulfate precipitate resembles the morphology of most protein particles more closely than the consistent, round shape of polystyrene beads. From left to right: barium sulfate, protein, and polystyrene beads.

Depiction of the size of visible particles in barium sulfate particle standard 3 (4 times magnification).
A set of seven barium sulfate particle standards composed of gradually increasing concentrations of barium sulfate was developed and qualified for a comparative assessment of small inherent particles in biopharmaceutical products. The method provides a tool to rank samples containing particles by comparing them side-by-side to the particle standards (1⇓⇓⇓⇓⇓–7) shown in Figure 4. The method provides an opportunity to move from a qualitative description of the amount of particles (e.g., “contains few particles”) or a plus system (e.g., “+” means few particles, “++” means more particles) to a better controlled, semi-quantitative method with qualified particle standards.

Barium sulfate visible particles standards (4 times magnification): (A) standard 1, (B) standard 3, (C) standard 5, (D) standard 7.
Materials and Methods
Materials
Barium sulfate and sodium azide were obtained from Sigma-Aldrich (St. Louis, MO). Water for injection (WFI) was purchased from Abbott (Abbott Park, IL). An Apollo II Liquid Viewer light box from Adelphi Manufacturing Co. Ltd. (West Sussex, UK) was used for visual inspection. A calibrated lux meter was used to periodically check the intensity of the light box.
Visual Inspection Method: Standardized Procedure
The visual inspection method is a subjective test, in which the instrument is the inspector's eye. The test can be affected by many factors, which can result in inconsistency in reported results. Some of these variables are type of light, intensity of illumination, sample handling, duration of observation, and inspector tiredness. Visual acuity of the inspector and personal differences in perceptions and interpretations are also very important factors causing variation. Control of these variables is essential to producing reliable results.
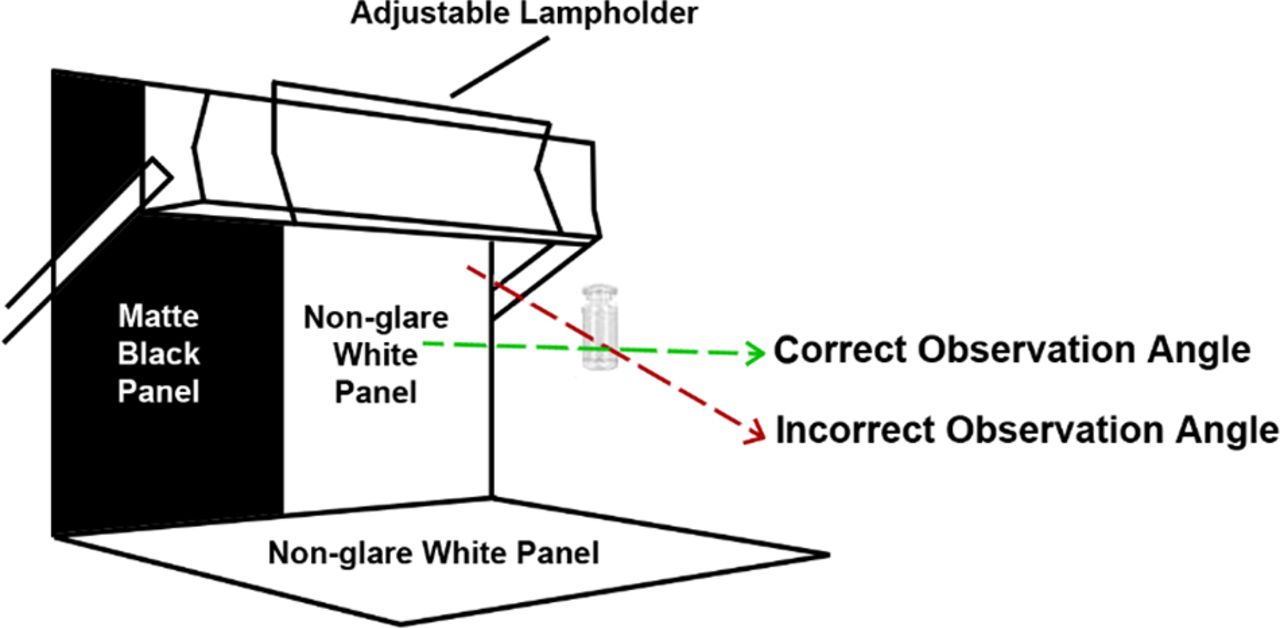
A visual inspection viewer with matt black panel, non-glare white panel, and adjustable lamp holder fixed with a suitable white-light source (fluorescent lamp) is recommended for visual assessment. The Ph. Eur. guideline recommends light intensity between 2000 and 3750 lux, although higher intensity may be used for colored glass or plastic containers (2). Narrowing the range of light intensity helped to reduce assay variability. It is essential to observe samples at the recommended light intensity, as the visibility of particles increases with increased light intensity. In order to further reduce variability, inspectors should be subject to annual eye exams as well as analyst qualification and periodic re-qualification.
Standardizing the viewing procedure—by controlling variables such as the light intensity, the angle of observation, the number of seconds the inspector views the vial (5–10 seconds is recommend), and period of time they should inspect—ensures that qualified inspectors will perform the test consistently. Figure 3 illustrates the correct angle of observation according to Medimmune procedures.

In the standardized inspection procedure using a light box with black and white background, the “correct” angle of observation was defined as 180° angle between an analyst and a test sample to minimize assay variability in the Medimmune procedure.
Preparation of Particle Standards
A set of particle standards for visual appearance testing was prepared as serial dilutions by pipetting from 0.2 to 0.001 mg/mL of barium sulfate in water for injection (WFI) containing sodium azide (addition of sodium azide is optional; it is added to eliminate bacterial growth by WFI), as shown in Table I. Standard 1 is most diluted and contains the least amount of particles. Standard 7 is the most concentrated suspension and contains the highest amount of particles (Figure 4). Barium sulfate particle standards were characterized by flow imaging and light obscuration methods. The amount of particles increased proportionally with increase of barium sulfate concentration for all particle size ranges, and the distribution of particle sizes was similar for all standards. These standards were visually shown to be stable for at least 3 months at 2–8 °C. After 6 months, the barium sulfate precipitate begins to climb up the wall of the vial and no longer simulates inherent protein particles typically found in high-concentration protein solutions.
Procedure for Preparation of Barium Sulfate Particle Standards
Inspection of Samples
Test samples are viewed side-by-side against particle standards using a light box with the black and white background as shown in Figure 3. Results of inspection are reported as the standard to which the sample most closely matches. If the sample falls between two standards, the results should be reported as being less than the standard containing the greater number of particles. For example, if a sample falls between standards 2 and 3, the result should be reported as <Standard 3. Particles in test samples and standards may settle and therefore should be gently swirled before use.
Result and Discussion
There is an industry-wide unmet need of particle standards for visual inspection of protein therapeutics. This need was satisfied by the development of barium sulfate particle standards that provide a tool to monitor and control small, inherent visible particles in biopharmaceutical products. The described method enables development of optimized protein formulations and allows monitoring of proteinaceous particles at product release and over the course of the product's shelf-life. The method uses various concentrations of barium sulfate particles in water that simulate the appearance of visible proteinaceous particles because of their irregular size and shape, as well as their heterogeneous particle size distribution, ranging from sub-micron up to visible (Figure 2). There is no universally accepted definition of the size of a visible particle. Most companies define particles as “visible” in the 100–200 μm size range based on the capability of detecting a single particle in a vial (9, 13). However, protein particles smaller than 100 microns may be seen during visible inspection. This method enables semi-quantitation of the small visible particles typically found in protein therapeutics. Particles in the barium sulfate suspension are smaller than 100 μm (Figure 5) and can also be characterized by sub-visible particle (SVP) analysis (Figure 2). Flow microscopy was used to get an estimate of the amount of particles in the barium sulfate particle standards. As barium sulfate concentration increases in the standards (from Standard 1 to 7), there was a proportional increase in the number of particles in all size ranges (≥2 μm, ≥10 μm, and ≥25 μm). A particle on the verge of visual acuity cannot be observed as a single particle in a vial. However, multiple particles can be visualized when observed in motion under sufficient light intensity. The light enables illumination of the particles (8), similar in principle as that of automated inspection machines. This method is not quantitative, as the exact number of protein particles cannot be determined. However, side-by-side comparison of the test sample with standards can provide semi-quantitative visible particle assessment and dispositioning. For lot release, semi-quantitative results can be reported and trended. The acceptance criteria can also include the standard limit that would define the qualitative specification (such as “some” protein particles may be present).
Setting Acceptance Criteria for Visible Particles
It is critical to set a maximum level of allowable proteinaceous particles in the drug product and ensure that level is not exceeded over the product's shelf-life. Protein therapeutic product specifications typically contain a qualitative description of visible particles such as “some or few proteinaceous particles may be present”. A semi-quantitative method that defines the word “some” or “few” is therefore desirable to make these terms more objective and capable of being applied consistently across the industry. The highest acceptable level of visible particles using this method in a biopharmaceutical product could be set using development and clinical data for that respective product. However, during the early stages of development, very limited data are available related to the particle formation in a specific product. In order to define “some” for these early stage products, the following studies were undertaken:
Visual assessment of five commercial products and 26 MedImmune clinical biopharmaceutical products were tested at various stages of their shelf-life (Figure 6). Results in Figure 6 show that most products contained inherent visible particles comparable to those levels seen in barium sulfate particle Standards 1 and 2. Some products had particles comparable to Standard 3 and 4. All 31 tested drug products, including commercial products, had inherent particles with level less than Standard 5. Using barium sulfate particle standards enabled quantification of the number of visible inherent particles in the products. The data clearly suggest that the use of this method provides a more accurate and robust monitoring tool for proteinaceous particles compared to testing visible particles without standards.
A study was conducted using clinicians, nurses, pharmacists, and scientists who were not qualified in the visual inspection method to identify the barium sulfate particle standard level (from Standard 1 to 7) that would exceed the maximum permissible level of inherent particles in drug products. The unqualified personnel were not able to see any particles in the particle standards below Standard 5 under visual inspection conditions recommended by the Ph. Eur. (2). Results obtained by the unqualified personnel can be considered representative of the inspection capability of patients and health care professionals who perform visual inspection of products prior to administration. However, trained, qualified personnel are able to see and quantitate these small inherent particles on the verge of visual acuity.
Based on the above results, the acceptable limit for inherent particles for clinical products was established as “less than Standard 5”. As development proceeds and more particle data are generated, the allowable limit for each product may be adjusted.
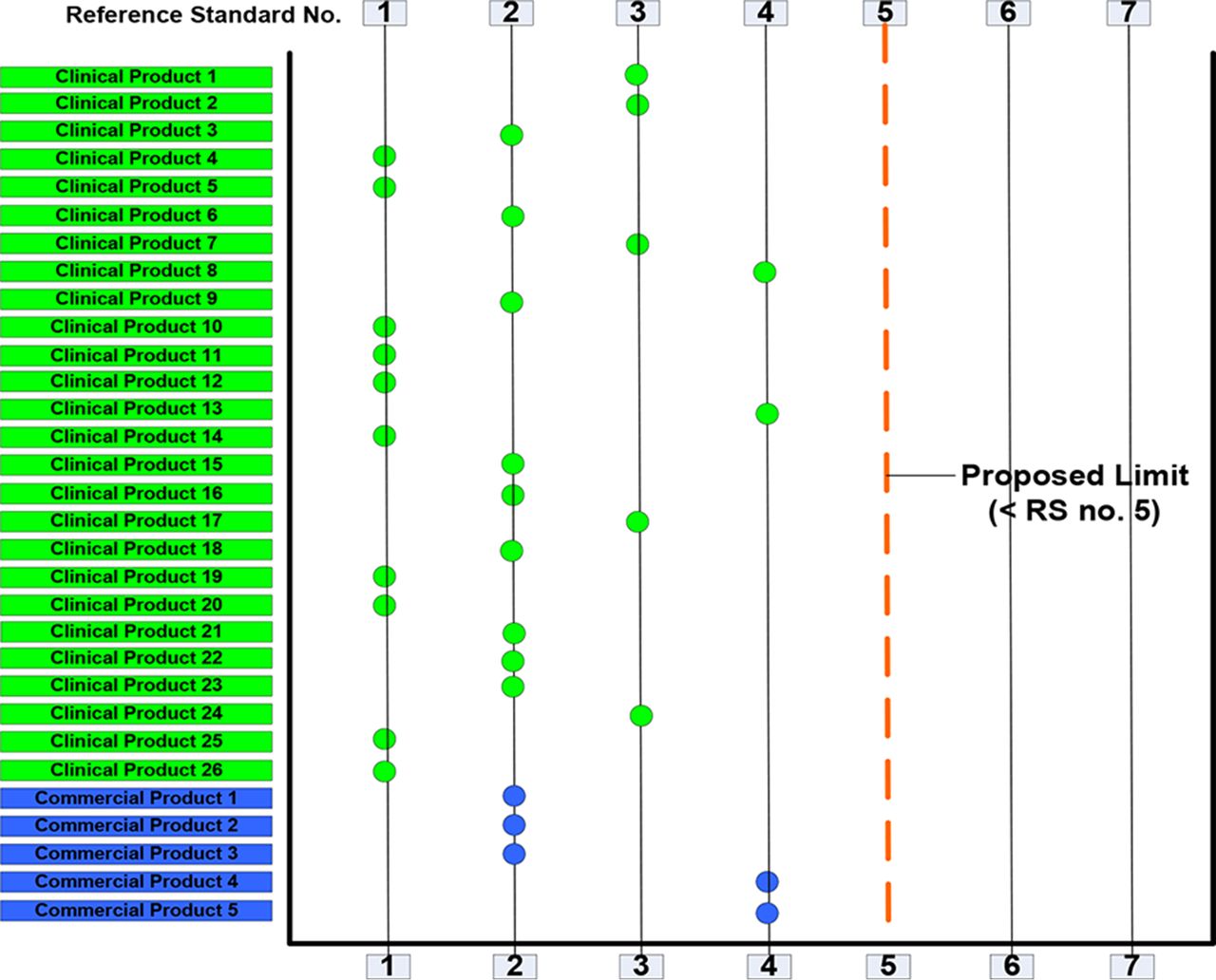
Results of visible particle assessment of clinical and commercial drug products.
Case Study
Application of Visible Particle Analysis in Formulation Development of a High-concentration Monoclonal Antibody (MAb) Product
A high-concentration MAb formulation was found to have increased propensity to form proteinaceous visible particles during stability (Figure 7). Formulation optimization studies were performed to evaluate the effect of two types of non-ionic surfactants (A and B) on various product quality attributes including visible particle and SVP formation. Visible particle analysis was performed using the method described in this paper. SVP analysis was performed using a micro-flow imaging instrument.

Visible particles in high-concentration MAb formulation without surfactant.
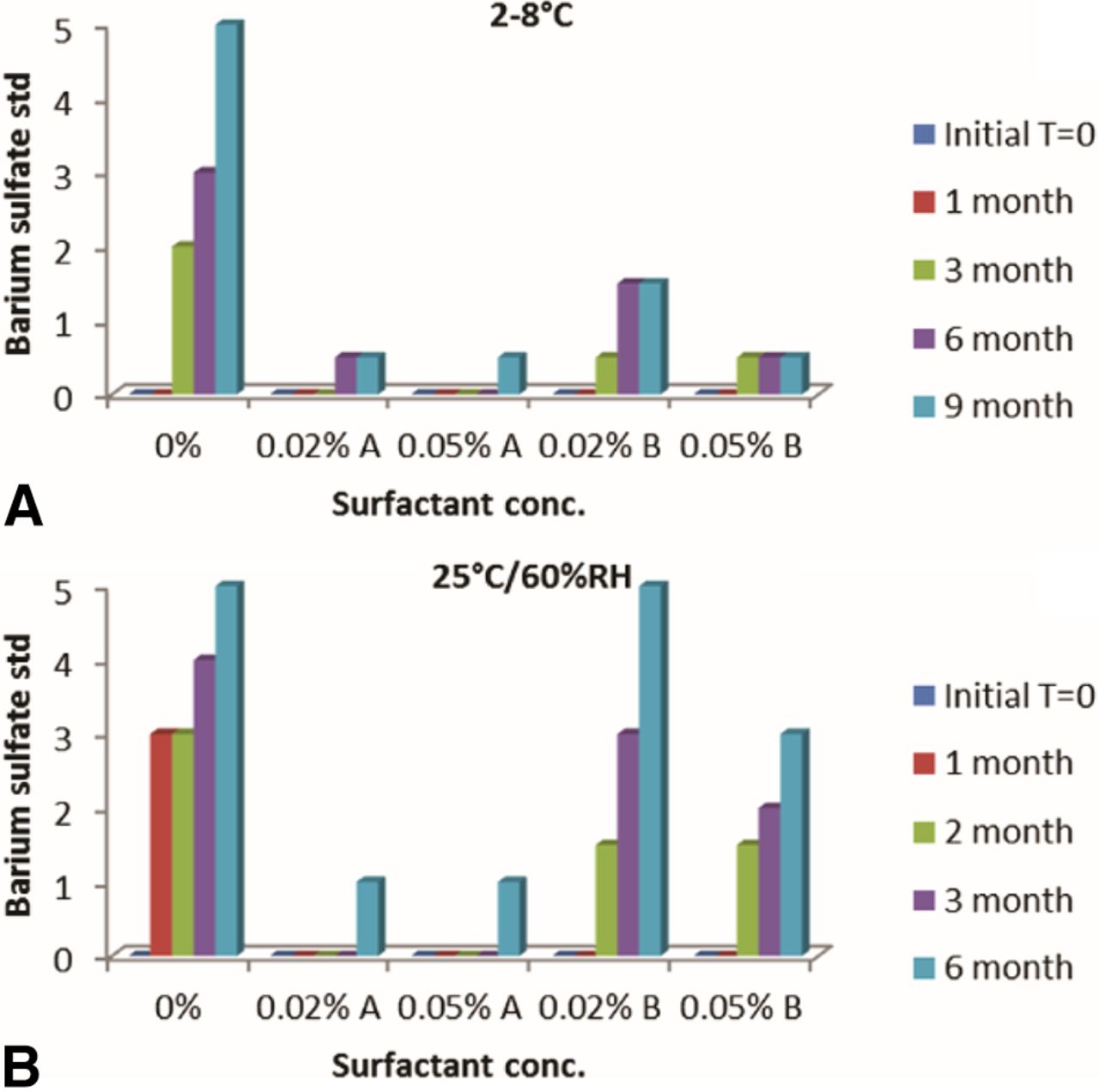
Formulation without surfactant demonstrated a significant increase in visible particles at 2–8 °C and 25 °C/60% relative humidity (RH). Formulation containing Surfactant A showed the least increase in visible particles on stability. Interestingly, Surfactant B was not as effective as Surfactant A in controlling the formation of visible particles. Also, in cases where visible particle formation increased in stability, it appeared to do so rapidly at 25 °C/60% RH compared to 2–8 °C (Figure 8). Differentiation between samples containing various levels of visible particles was made possible only through the use of barium sulfate particle standards.

Results of visible particle assessment using barium sulfate standards for MAb formulations on stability at (A) 2–8 °C and (B) 25 °C/60% RH conditions.
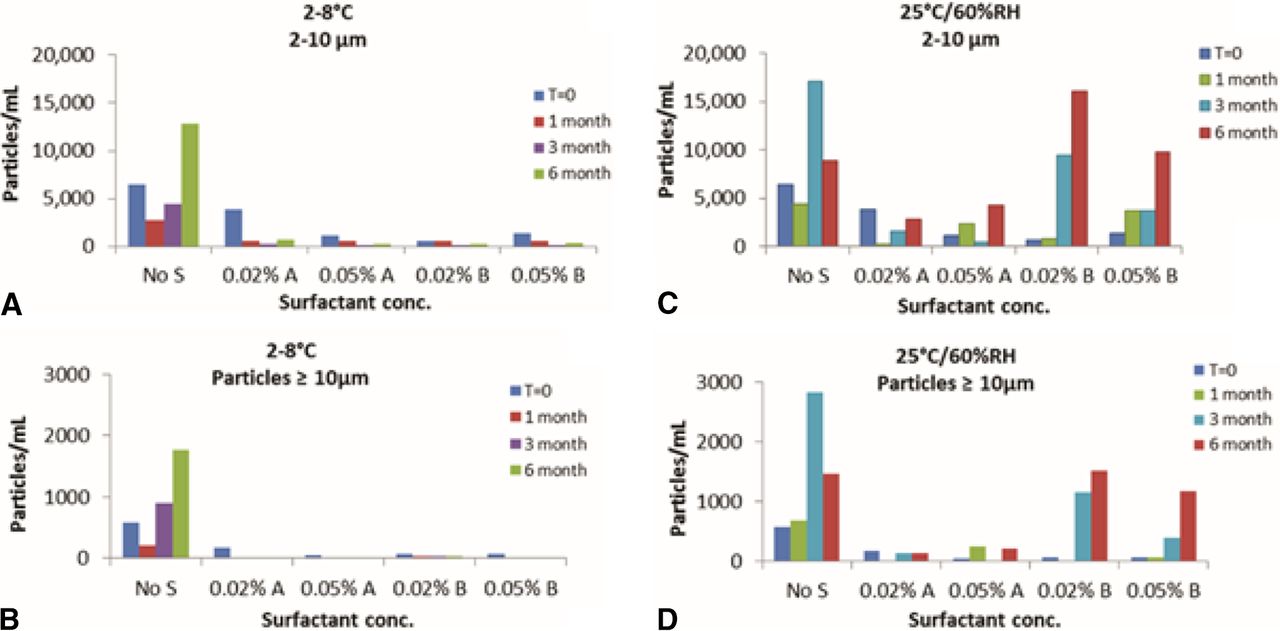
MAb formulation without surfactant had higher SVP counts (2–10 μm and ≥10 μm) compared to formulations containing surfactant. Formulations containing Surfactant A and formulations containing Surfactant B showed very low SVP counts after storage at 2–8 °C. However, after 3 months at 25 °C/60% RH, the formulation containing Surfactant B showed a significantly higher SVP count than the formulation containing Surfactant A (Figure 9). Increasing the concentration of surfactant (A or B) from 0.02% w/v to 0.05% w/v did not cause a significant change in visible particle or SVP count. This study showed a good correlation between SVP counts of protein particles and the semi-quantitative assessment of visible particles using the barium sulfate standards.

SVP analysis using micro-flow imaging for MAb formulations on stability at (A) 2–8 °C for 2–10 μm particles, (B) 2–8 °C for particles ≥10 μm, (C) 25 °C/60% RH for 2–10 μm particles, and (D) 25 °C/60% RH for particles ≥10 μm.
For the high-concentration MAb, visible particle analysis using barium sulfate standards was crucial for monitoring the effect of the growth of particles on stability. Results obtained led to the selection of an optimal formulation that minimized the formation of visible particles and SVPs.
Method Verification
The consistency of the semi-quantitative method was assessed by multiple analysts over time. Two trained inspectors routinely tested all clinical products side-by-side over the shelf life as part of the continuous method optimization. The samples were blinded so that the inspectors were not biased by previous observations. A total of 196 clinical product samples were tested and paired for inspector-to-inspector performance. In 27 out of 196 product samples, the difference between the two inspectors was one standard unit. In the remaining 169 product samples, the same results were obtained between the two inspectors. Implementation of barium sulfate standards made the visible inspection method much more objective, reducing analyst-to-analyst variability.
Conclusion
All protein solutions contain particles that vary greatly in visibility and size from invisible (sub-micron) to visible (millimeter) and, as part of their control strategy, biopharmaceutical companies are required to monitor and minimize the presence of visible particles and SVPs in their products. Most visual inspection methods are subjective and affected by the individual inspector's perception and interpretation and, thus, may be highly variable and poorly controlled. A semi-quantitative method to monitor the amount of proteinaceous particle formation against visible particle standards was developed. Analysts should be trained and qualified (using challenge sets) to visually distinguish between proteinaceous and common foreign particles using this method. Particles when observed need to be characterized thoroughly for identity, nature, morphology, and quantity using orthogonal techniques.
The method offers several process controls to minimize variation in the procedure such as harmonizing the angle of observation and distance from the light source. The method is semi-quantitative because it uses barium sulfate standards at various concentrations that are visually compared to the proteinaceous particles in the sample and because an assessment of the number of particles present in sample versus standard is quantitated.
The implementation of the barium sulfate standards has made the visual appearance method more sensitive, less subjective, and more reproducible. The method allows semi-quantitative monitoring and control of proteinaceous particles upon lot release and over the shelf-life of the product. Additionally, the method aids in developing a formulation that minimizes proteinaceous particles and in assessing the impact of process changes on particle formation in a semi-quantitative manner.
Conflict of Interest Declaration
The authors declare that they have no competing interests.
Acknowledgements
The authors would like to thank Mary-Ann Jackson, Anju Parmar, and Nitashree Shivaprasad for their help in developing this method; Yoen Joo Kim and Sonia Dragulin-Otto for analysis of barium sulfate standards; and Nancy Craighead, Mark Schenerman, Jared Bee, and Mike Washabaugh for their helpful review.
- © PDA, Inc. 2016




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}