
Abstract
In 2014, the members of the BioPhorum Operations Group (BPOG) produced a 100-page continued process verification case study, entitled “Continued Process Verification: An Industry Position Paper with Example Protocol”. This case study captures the thought processes involved in creating a continued process verification plan for a new product in response to the U.S. Food and Drug Administration's guidance on the subject introduced in 2011. In so doing, it provided the specific example of a plan developed for a new molecular antibody product based on the “A MAb Case Study” that preceded it in 2009.
This document provides a roadmap that draws on the content of the continued process verification case study to provide a step-by-step guide in a more accessible form, with reference to a process map of the product life cycle. It could be used as a basis for continued process verification implementation in a number of different scenarios:
For a single product and process;
For a single site;
To assist in the sharing of data monitoring responsibilities among sites;
To assist in establishing data monitoring agreements between a customer company and a contract manufacturing organization.
LAY ABSTRACT: The U.S. Food and Drug Administration issued guidance on the management of manufacturing processes designed to improve quality and control of drug products. This involved increased focus on regular monitoring of manufacturing processes, reporting of the results, and the taking of opportunities to improve. The guidance and practice associated with it is known as continued process verification. This paper summarizes good practice in responding to continued process verification guidance, gathered from subject matter experts in the biopharmaceutical industry.
Executive Summary
In 2014, the members of the BioPhorum Operations Group (BPOG) produced a 100-page continued process verification (CPV) case study, entitled “Continued Process Verification: An Industry Position Paper with Example Protocol” (1). This case study captures the thought processes involved in creating a CPV plan for a new product in response to the U.S. Food and Drug Administration's guidance on the subject introduced in 2011 (2). In so doing, it provided the specific example of a plan developed for a new molecular antibody product based on the “A MAb Case Study” that preceded it (3).
This document provides a roadmap that draws on the content of the CPV case study to provide a step-by-step guide in a more accessible form, with reference to a process map of the product life cycle. It could be used as a basis for CPV implementation in a number of different scenarios:
For a single product and process;
For a single site;
To assist in the sharing of data monitoring responsibilities among sites;
To assist in establishing data monitoring agreements between a customer company and a contract manufacturing organization.
Introduction
This document is designed to support the definition and implementation of continued process verification (CPV) plans for manufacturing operations relating to biopharmaceutical drug substances in response to the U.S. Food and Drug Administration (FDA) guidance provided in 2011 (2) and similar guidance from the European Medicines Agency (EMA) in 2014 (4). However, the concepts, steps, and elements presented are applicable beyond biopharmaceutical drug substances. It takes the form of step-wise recommendations, cross-referenced with a process flow diagram (PFD), that have been authored by subject matter experts from across the industry, under the facilitation services of the BioPhorum Operations Group (BPOG).
While the document contains elements of good practice, it should be recognized that practice varies across the industry. Implementation is the responsibility of individual companies taking a risk-based approach in creating their procedures (5).
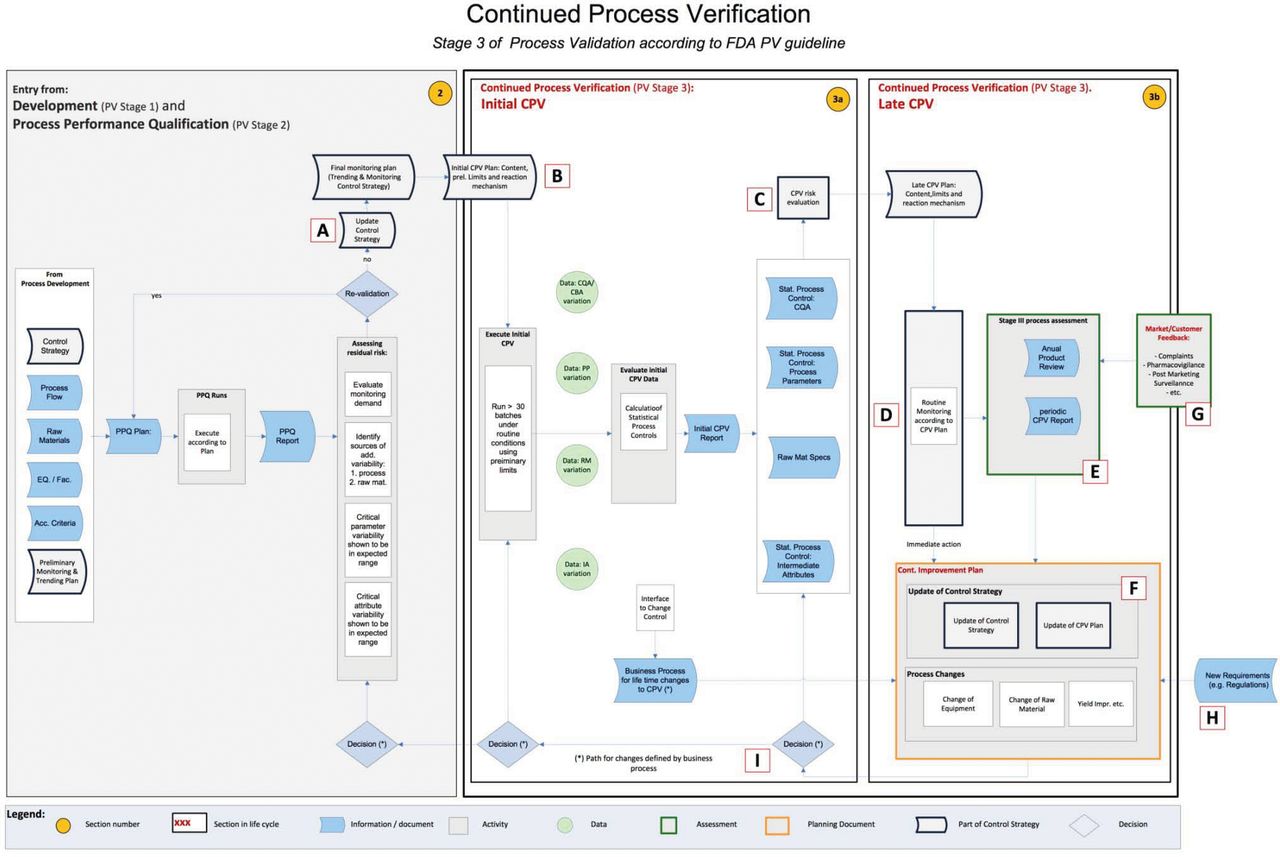
This document does not provide a detailed explanation of what CPV is, or attempt to motivate why it is important for companies to implement it. In brief, however, CPV is the proactive collection, analysis, and use of process performance and other data to demonstrate maintenance of control during the life cycle of a qualified process. It uses the principles of statistical process control and is useful to increase process understanding and decrease risk. The CPV system is an integral part of overall risk management and receives inputs from several other systems that support process design, development, qualification, non-conformance investigations, complaints, change controls, process data monitoring systems, and raw material testing. The CPV system outputs information to many other systems such as the annual product quality review, change control, and continuous improvement plans. Many of these interfaces are indicated in the accompanying PFD (see Figure 1).

A flow diagram showing the process of creating, operating and maintaining a continued process verification plan according to FDA guidance as interpreted by industry members of the BioPhorum Operations Group.
This document does not explicitly relate to the use of information technology (IT) systems, but the authors recognize such systems are an important factor in the efficient implementation of CPV plans. Ideally, highly efficient IT systems would be in a validated state, ready for use by the manufacturing operations function before manufacturing at the commercial scale takes place. Because this is not always practical, especially for legacy products, a manual CPV system may be introduced or an integrated IT system may be validated alongside a manual system over a period of time. It is entirely justifiable not to introduce an IT system if it is not going to be beneficial to the control of a particular production process. Companies are highly likely to evaluate the need for such systems on a product-by-product basis.
When writing this paper, the authors assumed that a number of pre-requisites would be in place:
A control strategy has been documented as part of development (Stage 1) and process performance qualification (PPQ) (Stage 2);
Some lots at commercial scale have already been manufactured, perhaps as part of PPQ;
CPV plans become active at the end of PPQ.
The steps in this roadmap are aligned with the FDA guideline Stages 1, 2, and 3 for the life cycle of a product. This document assumes that Stage 1 is complete and provides the basis for defining a CPV program during Stage 2 and eventually implementing it post-PPQ batches. The life cycle process steps are broken down into sub-stage phases and activities to aid understanding and implementation. Some content could be regarded as good practice, as it is commonly implemented across the industry, while other content is provided in the form of options for consideration in certain circumstances.
Process Validation Stage 2
Step 1: Collect Existing Documentation
At this stage, the key activity is the collection of existing documentation. The reader may wish to refer to the PFD in Figure 1, where this stage is marked “A”.
The objective is to understand what is already known about the process parameters and attributes that will need to be monitored as part of a CPV plan. Relevant documents may include development reports, control strategies, previous risk assessments, and assessments of critical quality attributes (CQAs), critical process parameters (CPPs), and critical material attributes.
For legacy products, that is, those that are already licensed, license section 3.2.S.2.4 contains important information about what control parameters and attributes agencies have already approved.
The relevant documents will ideally be co-located in a knowledge management system as an output from Stages 1 and 2 in the product life cycle. If documentation is not co-located and easily accessible, there may be an option to initiate or link into improvements in life cycle management.
Step 2: Collect Existing Process Data
Still at reference point A in the PFD, the purpose of collecting and examining existing process data is to enable the quantitation of historical process variation.
Existing data that are considered relevant to the current process should be gathered for all parameters and attributes likely to be part of the CPV plan (see Stage 3a, Step 1: Parameter and Attribute Selection). Data will likely come from process development, clinical, process qualification, and other full-scale production lots. These data will be used to confirm the parameters and attributes that will be included in the CPV plan and to establish preliminary CPV trend limits (see Stage 3a, Step 2: Determination of CPV Limits).
Wherever they are available, as entered, unrounded, or full precision data values should be used. Rounded data, which are often the reported or official results, are not preferred because they reduce the ability to accurately estimate variability in the system. This is true not only at this early stage of data collection, but also at all subsequent stages.
Similarly, values that are officially reported as <LOQ (less than the limit of quantitation) but that produce numeric results may be useful to trend. These values may be subject to greater uncertainty because they are less than an established LOQ; nonetheless, they can convey useful information about system performance when trended over time.
The collection of initial data offers the option to test existing systems for data collection, data aggregation, and data analysis. This exercise could be performed to assess the effort that will be required for ongoing CPV plan execution. If additional resources or electronic system improvements are required, this experience is likely to help in the justification of resources for the improvement effort.
Process Validation Stage 3
Stage 3 is usefully divided into 3a, referred to here as initial CPV, and 3b, referred to as late CPV.
Stage 3a: Initial CPV
Step 1: Parameter and Attribute Selection:
The initial CPV plan serves as the first documentation of the parameters and attributes to be monitored and the limits that will be used for monitoring. Providing justification for parameter and attribute inclusion or exclusion, as well as justification for the data and methods used for limit calculation, will make this document a powerful description of the company CPV strategy. Recent experience among companies represented suggests that this document is often requested by inspecting health authorities.
Parameter and Attribute Selection Based on Previous Assessments of Criticality:
One of the primary objectives in preparing the initial CPV plan is to create and justify the list of process parameters and attributes that will need to be tracked as part of an initial CPV plan. The assignment of criticality levels to individual parameters and attributes typically takes place at the end of process validation Stage 1 and may be refined through Stage 2. For legacy products, assignment of criticality may or may not have been completed prior to process validation Stage 3.
According to the most recent agency feedback, parameter and attribute criticality should be based on the potential of an operating parameter or raw material attribute to affect a CQA significantly. This classification should be independent of the relative occurrence or expected probability of such an excursion from specified limits happening. In other words, just because a parameter or attribute is well controlled, it cannot necessarily be deemed noncritical. However, from the standpoint of parameter and attribute selection for CPV, the controllability of a parameter or the capability of a process to control an attribute can be considered in the risk assessment. A stable and capable parameter or attribute may not need to be trended or could be trended at a reduced frequency. Parameters that are continuously monitored and controlled within specified ranges by automation systems (e.g., bioreactor temperature) are not typically included in standard statistical process control (SPC) charts. Alarms generated by the automation systems are often used to identify excursions of these parameters from specified limits.
The companies represented on this work agreed that at minimum, CPV plans should include a prioritized subset of critical process outputs such as CQAs. Companies may also be interested in intermediate attributes to promote an understanding of unit operations.
Parameter and Attribute Selection Based on Data:
In addition to selection of parameters and attributes based on established levels of criticality, historical data may also provide an indication of what parameters and attributes may be beneficial to include in a CPV plan. A review of data may indicate additional parameters and attributes that correlate with CQAs or are deemed important business measures. By definition, CPPs affect CQAs and are probably valuable to include in a CPV plan.
Where possible, looking at process capability indices (Cpks) or process performance indices (Ppks) (percent fall-out, etc.) of parameters and attributes may influence whether that parameter or attribute should be added to the CPV plan or not. However, these statistics are highly sensitive to sample size, which can be gauged by looking at a confidence interval on the index (6). It is always advisable to consult a qualified statistician if there is any concern about the sample size or the nature of the distribution of the data. Inclusion of parameters and attributes with low capability in the CPV plan will ensure that they continue to be tracked. Parameters and attributes with very low capability may deserve to be moved to a continuous improvement workstream as soon as practically possible. Parameters and attributes with high capability may not be valuable to monitor or could be monitored at a reduced level. Reduced monitoring would need to be described and justified in the CPV plan. Statistical guidance is suggested when using measures of process capability, especially when limited data underlie their calculation, to ensure these metrics are not over-interpreted.
Parameters and attributes that are not numerical can be excluded from statistical analysis. This might include attributes (color, etc.) or assays involving qualitative comparison to reference standards. Parameters and attributes that will not be measured on an ongoing basis should be excluded from the CPV plan, as they cannot be tracked over time (e.g., testing that was only performed as part of clinical or process qualification lots).
If a parameter or attribute has a limited number of unique values (pH controlled in a narrow range), it may not be statistically appropriate to calculate a mean and standard deviation, so these parameters or attributes should be excluded from statistical analysis. However, the company may want to track and visualize these results in some other way if the information is considered important.
Optimal Parameter and Attribute Set:
The optimal set of CPV parameters and attributes varies based on several factors unique to each product, process, and company; however, a risk-based approach is fundamental to selecting a valuable and defendable plan. A minimum set of critical parameters and attributes is expected to be monitored for compliance reasons; furthermore, maintaining visibility to additional parameters and attributes may provide greater insight into causes of process shifts and support process changes. Companies must balance compliance and expected business value against the required investment when scoping parameters and attributes for inclusion.
Some companies limit their scope of CPV by including only critical parameters and attributes in their formal CPV program, while taking a separate approach for non-critical parameters and attributes. Others may choose to monitor non-critical or business-related parameters and attributes through other process monitoring mechanisms and limit the scope of what is covered under the CPV plan. A wider range of parameters and attributes (including non-critical parameters and attributes) can be included in the CPV plan and these may be stratified based on criticality. Some companies have chosen to track several parameters and attributes through a certain number of lots that can later be evaluated for continued inclusion. For instance, companies may continue all measurements taken during PPQ through 30 lots, then revisit.
Step 2: Determination of CPV Trend Limits:
Moving to reference point B in the flow diagram, we start the process of determining the CPV trend limits and response mechanisms.
After collecting historical data and identifying the parameters and attributes that will be included in the monitoring plan, a preliminary CPV trend limit needs to be established for each applicable parameter or attribute. Statistically derived limits are preferable and are meant to represent the expected performance of the process in the future. These may be calculated based on several techniques, depending on the nature of the data. For instance, calculating tolerance intervals rather than 3-sigma limits may be a useful approach when data is limited.
Calculations are most straightforward for continuous, numerical data for which one point exists per manufacturing lot, such as final concentration, final product purity, or pH at a particular process point. Other types of data such as profile data, non-numeric results, and results with limited possible values are more complicated. In all cases, it is recommended to look at the nature and amount of data available and consult with a statistician to ensure calculated limits are justifiable.
In addition to direct data for the process in question, consider data from other sites and scales, data from similar processes, and development data to set preliminary CPV trend limits. Where a limited data set makes the calculation of statistical limits impractical, a scientifically or practically justified limit is more valuable than no limit during initial CPV, as it prompts action and revision as process experience increases. It may be seen as a risk to begin monitoring with preliminary CPV trend limits; however, doing so demonstrates an effort to control the process using the best knowledge available and should be understood by regulatory authorities. Even if CPV trend limits cannot be established, monitoring can start by simply charting the data in chronological order.
Reference Data Set:
Calculations of statistical limits must be based on some historical data set. Good practice would be to use all the available data that are considered representative of the current process. Changes, intended or unintended, may have taken place over time, which may make some data unsuitable for inclusion in calculations because they is no longer predictive of future performance. The representative data set usually consists of the most recent data and extends backward as long as the process has been in its current state, which may be termed the reference data set. Because some process changes affect some parameters or attributes while not affecting others, the reference data set may differ between parameters or attributes but should be defined and justified. Additionally, the raw data that comprise the reference data set should be documented to ensure traceability of all performed calculations.
Furthermore, in identifying the reference data set, data within the identified time frame may be stratified. For example, some batches may run in alternative equipment lines, at different scales, or with variable processing times. Factors such as these may produce data sets that are not statistically combinatory, despite representing manufacture of the same product.
Data should not be combined to establish a single set of CPV trend limits if they are generated on the basis of different underlying conditions. It may be best to plot these data on separate charts. Charting data separately may be valuable in terms of comparing process performance between suites or sites. Good practice is to include on all charts some type of standard so that there is the option to compare and combine data if required. Alternatively, it may be possible to standardize or normalize stratified data to enable the data to be plotted on one chart.
Number of Batches Required To Set Limits:
When setting preliminary CPV trend limits it is important to consider whether the product is produced in high or low volumes, the regularity of manufacture, or whether the product is made in campaigns. Typically, preliminary CPV trend limits are set if less than 30 data points are available. Companies may choose different threshold values, but 30 batches is a good rule of thumb.
For low-volume products, where generation of a significant number of batches will take years, an alternative approach is to establish a time period after which limit review or recalculation is required. For low-volume processes, judgment and justification will always be required.
Charting Data and Applying Rules:
Both the calculation of limits and the appropriate way of charting data are highly dependent on the nature of the data. For most numerical, continuous, one-value-per-lot data sets, simple run charts, control charts, and probability charts are most often used along with a check for normality.
Once data are visualized in an appropriate chart, runs rules are used to signal outliers, shifts, drifts, and trends within the data that may require evaluation. In this work, these are referred to as signals; alternatively, different companies may refer to these as statistical alerts, flags, rule violations, or statistical events. In practice, most companies use Shewhart control charts (7) with a subset of Western Electric rules or Nelson rules or a combination/variation of the two as statistical signals. Among the companies represented within BPOG, the following run rules were most often applied to the individuals chart:
1 data point outside mean ± 3 standard deviations (all companies used);
7, 8, or 9 consecutive points exclusively above or below the mean (used by most companies);
6 or 7 consecutive points trending exclusively upwards or downwards (not always used);
2 out of 3 consecutive points >2 standard deviations away from the mean on the same side (not always used).
A risk-based approach should be used to establish which charts and rules will be used. In most cases, it is advisable to consult a statistician. For example, when data departs from normality, consideration should be given to transforming the data. However, we should remember that we are seeking indications of trends and that it may be best to keep the data presentation systems simple in the workplace.
An internal guidance document that describes how statistical limits should be calculated and how data should be charted and evaluated will help to ensure continuity over time within a company. This document can then be referenced in other CPV plans or procedures.
Data System Design:
When developing the initial plan, consider how data are collected (manual, electronic, automated) and whether appropriate resources have been assigned to complete this collection and analysis. In all cases, the system for data collection, analysis, and reporting should ensure data integrity.
If an IT system is being used, it would ideally have been delivered by Stage 3a, though this is not always the case. Pragmatically, a start may be made and additional data sources and automation may be added over time through change management.
Step 3: Responses to Statistical Signals:
Statistical signals require some type of documented response in both initial and late CPV phases. However, the response to statistical signals during initial CPV, when preliminary CPV trend limits are in place, may not be as formal as when long-term CPV trend limits are in place. The response procedures for initial CPV should be described in the CPV plan or associated procedures.
Step 4: Recalculation of Limits:
Once a sufficient number of lots have been manufactured using the commercial process, the CPV program should be transitioned from initial CPV to late CPV. This transition is suggested at reference point C in the PFD. Thirty lots are typically considered sufficient for this transition; however, other factors should be considered, such as long-term sources of variation and manufacturing frequency. For example, if a product has a high frequency of manufacture, the first 30 lots may not capture all sources of common cause variation. In general, the transition from Stage 3a to 3b occurs as an event; however, all parameters and attributes may not transition at the same time due to changes that may affect a subset of the parameters or attributes. Parameter and attribute selection may be revisited based on additional process understanding gained during initial CPV, and addition or subtraction of parameters or attributes should be justified. Statistical limits should be recalculated based on the expanded reference data set. Statistical techniques used should be verified based on the data distributions in hand. CPV trend limits are now expected to remain the same as long as the process continues to perform in the same way. However, in practice CPV trend limits will need to change as other changes happen and may need to be periodically evaluated to ensure they remain predictive of future performance while taking care not to hide long-term process drift.
Stage 3b: Late CPV
Late CPV takes the general form of routine monitoring according to the CPV plan.
Step 1: Responses to Statistical Signals:
At point D in the PFD, we see another significant phase of the development of the CPV plan. At this point, routine monitoring commences in accordance with the plan and the organization starts to respond to the corresponding statistical signals.
Once a plan for CPV execution is established, business value is created in the execution of the plan. Business processes should be established in a procedure or plan that describes who does the monitoring and analysis, and how often. How are the analyzed results viewed, and by whom? What are the required responses to signals and who will execute them? Where are responses documented and when?
Evaluation of Statistical Signals:
A statistical signal is an indication that unexpected variation has been detected in the process that is statistically inconsistent. In other words, it is in violation of a statistical rule for a parameter or attribute being tracked in a CPV plan. Signals can result from real-time trending of the process or may be discovered after a periodic review of data. A procedure should be established that describes how the company evaluates and documents the cause of the signal. These evaluations would be similar to investigations conducted for non-conformances but may have important differences. For instance, because a statistical signal does not represent an excursion beyond specifications, it should not, in itself, prevent lot disposition. Companies may also choose to reduce the requirements for root cause determination associated with a signal evaluation. Additionally, Cpk or Ppk levels may be used to help decide whether signal evaluations are likely to be value added for specific parameters or attributes.
A risk-based process should also exist that will escalate the significance of a signal if product quality is potentially affected. This escalation may take the form of a signal evaluation converting to a formal non-conformance investigation that would need resolution prior to lot disposition.
Documentation of Statistical Signals:
While signal evaluations are similar to non-conformance investigations, they may be documented differently because they are indications of process inconsistencies, not necessarily of product quality risk. When asked what options exist for systematically documenting evaluations, the companies represented here responded as follows:
In most cases, an escalation procedure is put in place on a risk basis to identify what events need to be included in the quality non-conformance system. Events that are not escalated are still logged, evaluated, and discussed in CPV reports.
In some cases, companies implement a means of recording all events in their quality systems but handle signal evaluations within a different category that does not hold up batch release.
As is the goal with all evaluations, determination of the root cause of process variation should feed back into a continuous improvement mechanism. Corrective and preventative actions can result from a signal evaluation, and companies will need to determine how these actions are documented and tracked.
Step 2: Establish Real-Time Data Reporting:
The results of an ongoing monitoring effort may be most impactful if they are reported to interested parties as close to real time as possible. This type of reporting may be daily or weekly, for example. Clearly, electronic systems may enable this capability.
It is recommended that CPV data are made available to those working on the production floor. The operations or manufacturing teams will benefit from knowing if the process is in or out of control. Ideally, these data should be accessible soon after they are generated. To that end, charts available to operational staff may be refreshed quite frequently or automatically where electronic systems have been put in place to accomplish this. It is recommended that the operating personnel are also involved in discussions related to data interpretation, as they are most likely to understand and remember what happened in the process.
Where real-time verification of data integrity is not possible, companies should consider how to make data available as quickly as possible. Value can still be created even if data are trended before they have been audited/verified because the sooner the data are reviewed by operations and other process experts, the greater the likelihood relevant circumstances will be captured. Quality systems may need to be constructed to prevent the initiation of evaluations based on data that have not been audited/verified. To capture the value of early visibility, unverified data can be marked to indicate the status and prevent overreaction to data errors.
Step 3: Establish Periodic Summary Reports:
See point E in the PFD.
In addition to real-time reporting of process monitoring data, periodic summaries of CPV activities are considered good practice. A business process should be established that addresses how often the results of the CPV program are reported, in what way the reporting is accomplished, and who is the target audience. For example, formal CPV summary reports might be written on a quarterly basis to include all the observations made for a particular product.
Many of the companies represented in this work are using periodic CPV summary reports to help construct the formal annual product quality review required by some regulatory agencies.
Step 4: Revision of the CPV Plan:
Periodically, or by need, the CPV plan may be revised. This activity is shown at point F in the PFD. The activity also includes process changes motivated by the outputs of the CPV process.
The CPV plan should be a controlled document that is changed through a designated approval process. Changes to the CPV plan may result when previous assumptions are negated. Examples may include observation of raw material variability greater than that seen in the reference data set, or implementation of a process change, which shifts data to a new mean or modifies variability. In these examples, revised statistical limits and centerline would be required to be predictive of future behavior.
The CPV plan will require periodic revision such as when enough data are generated to replace preliminary limits with long-term limits as the plan transitions from initial CPV to late CPV. As process data accumulate, there may be reasons to remove or add parameters or attributes in the CPV plan. A mechanism for justifying these changes should be included in the change management process.
The policy for recalculating CPV trend limits needs to be documented. Rather than performing time-driven updates to CPV trend limits, which may lead to undetected process drift, established CPV trend limits are ordinarily not changed if the process has not changed and the overall behavior of a particular parameter or attribute continues as predicted. However, significant process changes will necessitate revision of CPV trend limits, and a new set of reference lots may be used to recalculate limits. Revised limits may return to a preliminary state until enough data have been collected in the revised process to set long-term limits again.
In addition to the need to update the CPV plan, learning gathered through the CPV process may lead to the need to make a manufacturing process or testing changes. These changes would follow established procedures for change control and should consider whether they require redevelopment or revalidation of portions of the manufacturing or testing process. These changes should also consider the need to change the CPV plan in parallel.
While process improvements are taking place, it may be desirable to remove some requirements for evaluation of signals from some parameters or attributes with known robustness issues. Such parameters or attributes would typically continue to be monitored while improvement is taking place. However, because the signals may be regular and expected, provision may be desirable within the procedures for signal evaluation and reporting requirements to be eased.
Step 5: Expansion of Inputs:
As shown at point G in the PFD, there may be drivers to extend CPV.
The aspirations for CPV in many companies extend beyond monitoring of process performance variables. Indeed, FDA guidance on the subject makes reference to several other potential sources of information, which may be helpful in understanding and controlling a manufacturing process, such as “defect complaints, out-of-specification findings, process deviation reports, process yield variations, batch records, incoming raw material records, and adverse event reports” (2). While quality systems exist to monitor each of these things, existing systems will have often been established separate from each other and information may not easily pass from one system to another. For instance, raw materials may be tested at the vendor as a requirement of receipt, but test results may not be easily used in conjunction with process performance data to understand and control the variability resulting from that raw material. Great value may exist in aggregating many existing sources of data, but additional IT and human resource challenges arise as the scope of the CPV effort broadens. Most of the companies represented in this work have taken or are taking a stepwise approach by establishing monitoring of core data, and later expanding to include other potentially valuable data sources.
Step 6: Regulatory Interactions:
Reference H in the PFD shows this area of the CPV process. Increasingly, regulatory agencies will come to expect robust CPV programs rooted in sound process understanding. Agencies may also come to expect quality metrics derived from the CPV program to be presented in annual reviews, filed changes/variations, or inspections. Inspection frequency may be influenced by the quality of a company's CPV system and reported metrics for process health.
As reviewers and inspectors become more familiar with the information available in a good CPV program, it is also anticipated that the CPV program could be leveraged to justify the process impact of changes, or to reduce the upfront validation requirements associated with changes. Companies should also be able to allay regulatory concerns about “white space” within specifications because an additional layer of process control exists associated with the CPV program.
The ability to leverage CPV plans during the life cycle of a product may be enhanced by sharing CPV plans with agencies during product review, not just site inspection. It has been reported that CPV plans, and related process life cycle management plans, have already been included by some companies in regulatory approval applications.
As CPV is constantly capturing critical information whether a process is remaining in a state of control, these data could be used to justify a risk- and fact-based approach to revalidation (reference I in the PFD). During the process life cycle, accumulated critical process information and the quality of supporting data could be used to justify different levels of activities (e.g., update CPV plan, change control strategy, go back to early CPV, or revalidate process).
Conclusions
The process outlined in this paper represents a general recommendation for implementation of CPV for new or legacy products. These high-level steps can be useful in driving alignment of understanding across the biopharmaceutical industry. The steps could also be used within a company or among companies to aid in the planning and implementation of a CPV program. A risk- and fact-based approach to process revalidation based on CPV data can support focusing effort on value-adding and quality-improving activities. Implementation of a CPV program is a current compliance expectation and is expected to influence regulatory inspections and reviews to an increasing degree in the future. Beyond compliance, significant business value can be created through implementing CPV systems that highlight risk, motivate improvement, and document increases in process reliability and performance.
Conflict of Interest Declaration
The authors declare there are no financial or non-financial competing interests related to this article.
Abbreviations
Glossary
Acknowledgments
Bert Frohlich (Shire), Eric Hamann (Pfizer), Mark DiMartino (Amgen), Martha Rogers (Abbvie), Robert Grassi (Lonza), Parag Shah (Roche), and Willy Ribus (Janssen), and the extended BPOG membership.
Footnotes
DISCLAIMER: The following article is a special editorial contribution from the BioPhorum Operations Group (BPOG). Please note that it did not go through the PDA Journal of Pharmaceutical Science and Technology regular peer review process.
- © PDA, Inc. 2016




{kind=link}