
Abstract
Terminal sterilization is considered the preferred means for the production of sterile drug products because it affords enhanced safety for the patient as the formulation is filled into its final container, sealed, and sterilized. Despite the obvious patient benefits, the use of terminal sterilization is artificially constrained by unreasonable expectations for the minimum time-temperature process to be used. The core misunderstanding with terminal sterilization is a fixation that destruction of a high population of a resistant biological indicator is required. The origin of this misconception is unclear, but it has resulted in sterilization conditions that are extremely harsh (15 min at 121 °C, of F0 > 8 min), which limit the use of terminal sterilization to extremely heat-stable formulations. These articles outline the artificial nature of the process constraints and describe a scientifically sound means to expand the use of terminal sterilization by identifying the correct process goal—destruction of the bioburden present in the container prior to sterilization. Recognition that the true intention is bioburden destruction in routine products allows for the use of reduced conditions (lower temperatures, shorter process dwell, or both) without added patient risk. By focusing attention on the correct process target, lower time-temperature conditions can be used to expand the use of terminal sterilization to products unable to withstand the harsh conditions that have been mistakenly applied. The first article provides the background and describes the benefits to patient, producer, and regulator. The second article includes validation and operational advice that can be used in the implementation.
LAY ABSTRACT: Terminal sterilization is considered the preferred means for the production of sterile drug products because it affords enhanced safety for the patient as the formulation is filled into its final container, sealed, and sterilized. Despite the obvious patient benefits, the use of terminal sterilization is artificially constrained by unreasonable expectations for the minimum time-temperature process to be used. These articles outline the artificial nature of the process constraints and describe a scientifically sound means to expand the use of terminal sterilization by identifying the correct process goal—destruction of the bioburden present in the container prior to sterilization. By focusing attention on the correct process target, lower time-temperature conditions can be used to expand the use of terminal sterilization to products unable to withstand the harsh conditions that have been mistakenly applied. The first article provides the background, and describes the benefits to patient, producer, and regulator. The second article includes validation and operational advice that can be used in the implementation.
- Terminal sterilization
- Aseptic processing
- Sterilization
- Biological indicator
- Bioburden
- Probability of a non-sterile unit (PNSU)
- Regulation
- Sterility assurance
The pharmaceutical industry succeeds by satisfying demands for products that will improve patient outcomes by filling gaps between available therapies. That's been accomplished by the introduction of new chemical or biological entities, novel dosage forms, and innovative delivery systems. Therapeutic gaps are filled by the evidence-based, case-by-case evaluation of new drugs, biologics, and drug delivery systems. It is time for the industry and those who regulate it to create an environment in which comparable levels of innovation are possible in sterile product manufacturing. Unfortunately, rigid, prescriptive regulations in drug manufacturing have a negative effect on innovation and, as a result, gaps between what is compliant and what is technically possible continue to grow.
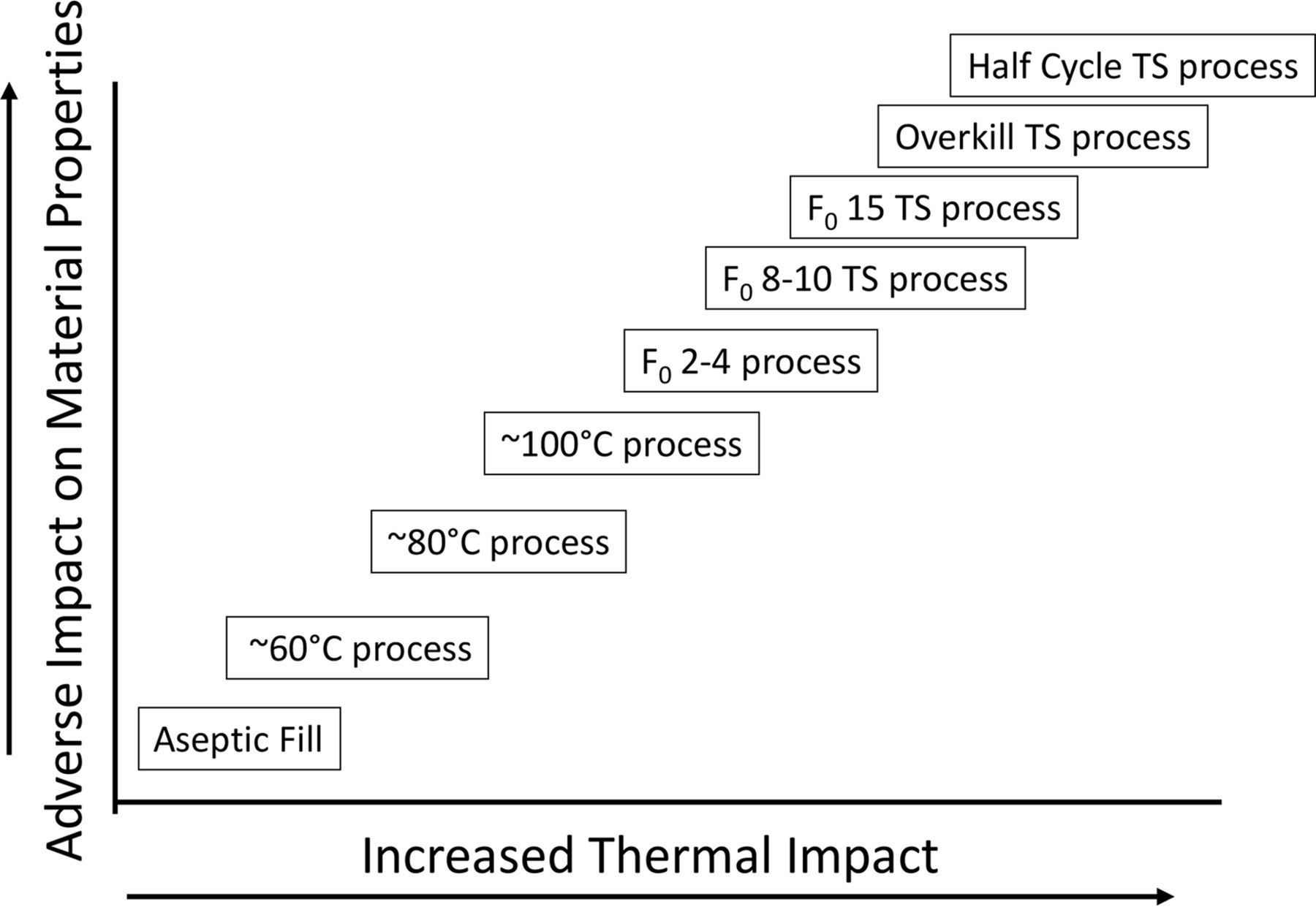
A gap of a different sort has existed since the first sterile drugs were prepared (see Figure 1).

Sterile processing gap.
This gap is the obvious distinction between sterile products that are aseptically produced (AP) and those that can be terminally sterilized (TS). The gap between these alternatives has resulted in divergent production methods. The demonstrable certainty of safety afforded by terminal treatments has been supported by regulatory preferences for their use. It follows logically that if AP is safe, terminal processes operating within the gap would be potentially safer still with the additional benefit of being easier to consistently control.
Unfortunately, regulatory guidance documents have caused confusion by simultaneously mandating higher-than-necessary TS process conditions while also recommending increased use of TS (1⇓–3). As a result the accessibility of terminal processing, which should be increasing, has actually decreased. With an increasing diversity of means for the production of sterile products available, the pharmaceutical industry has employed some newer concepts, but sadly perspective, unwisely conservative, and inflexibly regulation has stifled optimization of both AP and TS processes. Aseptic processing has evolved further, with isolation technology representing the current state of the art. However, the implementation of advanced aseptic processing has been slowed by at least two decades by unscientific and overly prescriptive regulation.
Terminal sterilization has undergone fewer technological changes, with the major changes relating to the size of the sterilizers. The distinction between the processes has sharpened with increasing use of large molecular therapies incapable of withstanding the stresses of thermal and radiation processes.
The choice of either an aseptic or terminal process has existed for many years, and the global pharmaceutical regulatory compliance environment has slowed innovation in both AP and TS. These failures are in and of themselves unfortunate, but the failure to allow processing innovators to explore the gap between these technologies is arguably even more problematic. A brief history of the salient activities can be useful in understanding the current situation.
In the 1940s, the USP included a sterilization expectation of 250 °F (the metric conversion of which is 121 °C) in USP XII for sterilization, and that temperature (and at times an exposure period of 15 min) has appeared in numerous USP chapters to this day (2). This expectation was removed from the media preparation and general sterilization content of USP starting in 2013 as the arbitrariness of the fixed process expectation was realized; however, numerous other mentions of these conditions remain within USP chapters (3).
In 1976, The U.S. Food and Drug Administration (FDA) issued a draft regulation, Current Good Manufacturing Practice in the Manufacture, Processing, Packaging or Holding of Large Volume Parenterals (1). This regulatory proposal (which never became a final regulation) includes numerous recommendations that have shaped sterilization practices. One of the less noticed sections of the 1976 draft is 212.5 (d), which described how sterilization processes with an F0 of less than 8 min could be accepted. The intimation in 1976 was that any process with an F0 greater than 8 min could be considered overkill.
An explicit preference for the use of terminal sterilization was stated by the FDA in 1991 in a proposed draft change to current good manufacturing practice (cGMP) regulations (4). The FDA was concerned at that time about the use of AP to manufacture generic versions of products that had been terminally sterilized by their innovators. The initiative sought to mandate increased usage of terminal sterilization. Industry, and most prominently the Parenteral Drug Association (PDA), while receptive to the intended goal, requested clarification on the specific terminal process to be used (5, 6). Prominent among the PDA comments to the proposed regulation was the suggestion that sterilization be considered with greater flexibility: “A narrow interpretation of terminal sterilization could in effect serve to reduce the use of post-aseptic fill sterilization processes rather than encourage their utilization.”
The European Medicines Agency (EMA) approached the subject in 1999 from a very different perspective by stating a preference for a 121 °C for 15 min process, with a fallback proposal for a minimum F0 of 8 min with a sterility assurance level (SAL) of 10−6 (7). This document has largely shaped contemporary western European and U.S. practices with regard to terminal sterilization. The Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme (PIC/S) ignored comments by industry subject matter experts who argued against such a narrow, rigid, and prescriptive TS process requirement.
There have been some efforts to consider the gap between TS and AP, and some have enjoyed a measure of success. The FDA first issued a concept paper on aseptic processing in 2002 prior to updating its guidance (8). That draft included the following content: “It is a well-accepted principle that sterile drugs should be manufactured by aseptic processing only when terminal sterilization is not feasible. However, unacceptable degradation of the product can occur as a result of terminal sterilization, or the market presentation can afford some unique and substantial clinical advantage not possible if terminal sterilization were employed. In such cases, adjunct processing steps (e.g., heat exposure conditions which provide some F0) to increase the level of sterility confidence should be considered.”
This section brought new attention to the subject, and a Product Quality Research Institute (PQRI) subgroup consisting of both FDA and industry was formed to discuss the concept paper in greater detail (9). It was agreed that while the objective for flexibility in TS was laudable, its inclusion in an aseptic processing guidance was not easily accomplished, as there were a number of technical issues to be resolved. There was consensus opinion to develop this subject independent of the AP guidance. The 2003 draft and the final version of FDA's guidance treated the subject in a less definitive manner, including only the first sentence from the 2002 concept paper (10, 11). The output of the previously mentioned PQRI recommended activity was apparently expected to address the subject of TS more fully. However, no efforts to discuss the matter of the gap between TS and AP were ever initiated as an outcome of the PQRI recommendation.
Japan took the next steps in addressing terminal sterilization processes with a comprehensive treatise on the subject (12). Included in the document was a broader interpretation of processing expectation with no mention of specific processing conditions but an expectation that “a sterility assurance level should be less than 10−6.” Significantly, this document re-introduced the concept of low F0 processes, which PIC/S had excluded from consideration in 1999: “If sterilization of pharmaceutical products at F0 >8 minutes is not feasible because of low heat tolerance of the formulation or containers, alternative sterilization conditions should be established by identifying process parameters to ensure a SAL of less than 10−6 and … Sterilization conditions thus established should be demonstrated to be scientifically valid.”
This very comprehensive document goes on to describe terminal sterilization process where F0 and time-temperature do not attain the PIC/S suggested minimum F0. This document has not had much impact outside of Japan, which is unfortunate in that it approaches the subject of terminal sterilization in a rigorous, patient-centered, and pragmatic manner and clearly allowed for a broader implementation of TS. The success of Japan's approach is manifested in not only the numbers of products sterilized in Japan using TS in a more flexible manner, but also their record of absolute patient safety. It should be noted that Japan currently uses low temperature moist heat extensively and experiences little difficulty with this use (13).
Following a series of sterilization conferences in 2009, a PDA task force was organized to develop industry guidance on post-aseptic fill lethal treatments. Achieving consensus proved harder than expected. While some participants believed these processes should be considered sterilization; others considered them as something less certain. After considerable discussion, a mutually acceptable draft was published that did not resolve the core dispute (14). The culmination of the task force effort included the following recommendations:
“The spectrum of post-aseptic fill sterilization treatments would eliminate the current ‘processing void’ and could include:
Steam Processes
60 °C, 80 °C, 100 °C processes (temperatures are arbitrarily chosen).
Low F0 processes (less than 8 minutes) and accomplished at less than 121 °C.
Radiation Processes
Processes at less than 15 kGy (minimum possible using VDMAX dose setting).”
This message was repeated and expanded upon in a recent publication, but regulators have largely ignored the process gap and have made little effort to close it (15).
In April 2016, the EMA issued a draft revision of its 1999 document entitled, Guideline on the Sterilisation of the Medicinal Product, Active Substance, Excipient and Primary Container (2). This revision included a suggestion for a microbial reduction process defined as “treatment at conditions that provide a lower lethality than sterilisation.” This definition lacks the necessary specificity to be broadly useful—its upper limit is an arbitrary F0 of 8 min and its lower limit undefined.
A New Understanding
Closing the gap requires changing the core paradigms that have plagued sterilization for decades. There are two essential questions: First, what exactly is a sterilization process expected to do, and second, how is that sterilization process to be evaluated? Addressing these essential questions can lead to a direct means to close the process gap.
What is the Sterilization Process Expected To Do?
A sterilization process must destroy the microorganisms present in/on the material in order render it safe for use and that means delivering a product incapable of producing an infection in the patient. Those microorganisms should be recognized as the natural bioburden in/on the materials and not the biological indicator (BI) used in the validation effort.
The BI serves as a measurement tool for the process and should never be used to define its duration. BIs by virtue of their increased and consistent resistance to the sterilization process provide a convenient means to determine that the physically measured lethality roughly correlates to a biological effect.
BIs can simplify validation when they are used in the brute force manner often somewhat misleadingly called overkill. Using them in overkill situations allows the end user to largely ignore the bioburden population and resistance. This approach results in a sterilization process that delivers lethality well beyond that necessary to ensure microbiological safety and should only be considered necessary with materials impervious to the adverse effects of intensive thermal exposure.
The real target of the sterilization process, however, is the bioburden microorganisms as they are present during routine sterilization and whose destruction must be assured on a daily basis. As radical as it might seem to those who do not possess the necessary understanding to be involved in decision making regarding microbiological safety, destruction of the BI during the validation is not a meaningful requirement. Its survival reveals more about the process performance than its death. Neither are achieving temperatures of 121 °C or killing BIs with a population of a million spores. Pre-sterilization bioburden is generally less than 1000 colony-forming units (cfu), and when aseptic processing is used for filling it is substantially lower, essentially nil. The resistance of bioburden subsequent to aseptic processing is that of vegetative cells, having virtually no resistance to heat (none of the common human, environmental, or disease-causing bacteria survives more than a few minutes at 70 °C).
How Is the Sterilization Process To Be Evaluated?
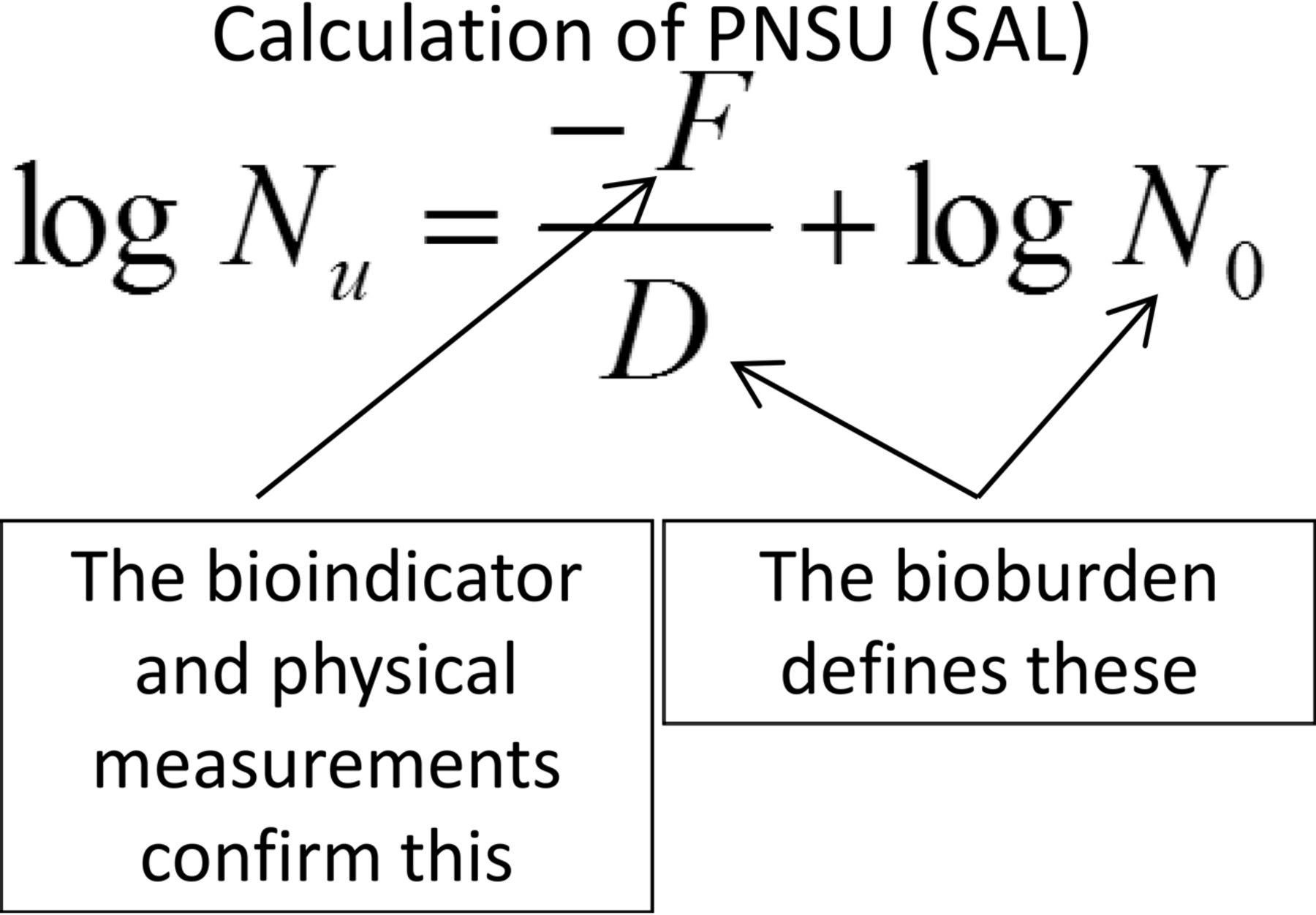
If the true goal in sterilization is the destruction of the bioburden present, then the probability of a non-sterile unit (PNSU) must be not greater than 1 unit in 1,000,000 units, as this is the minimum process expectation. The population and resistance of the bioburden should be known (or at least estimated) at the conditions of the sterilization process to determine the PNSU. This understanding was reintroduced by USP in <1229> Sterilization (5). The means for calculation of PNSU are shown in Figure 2.
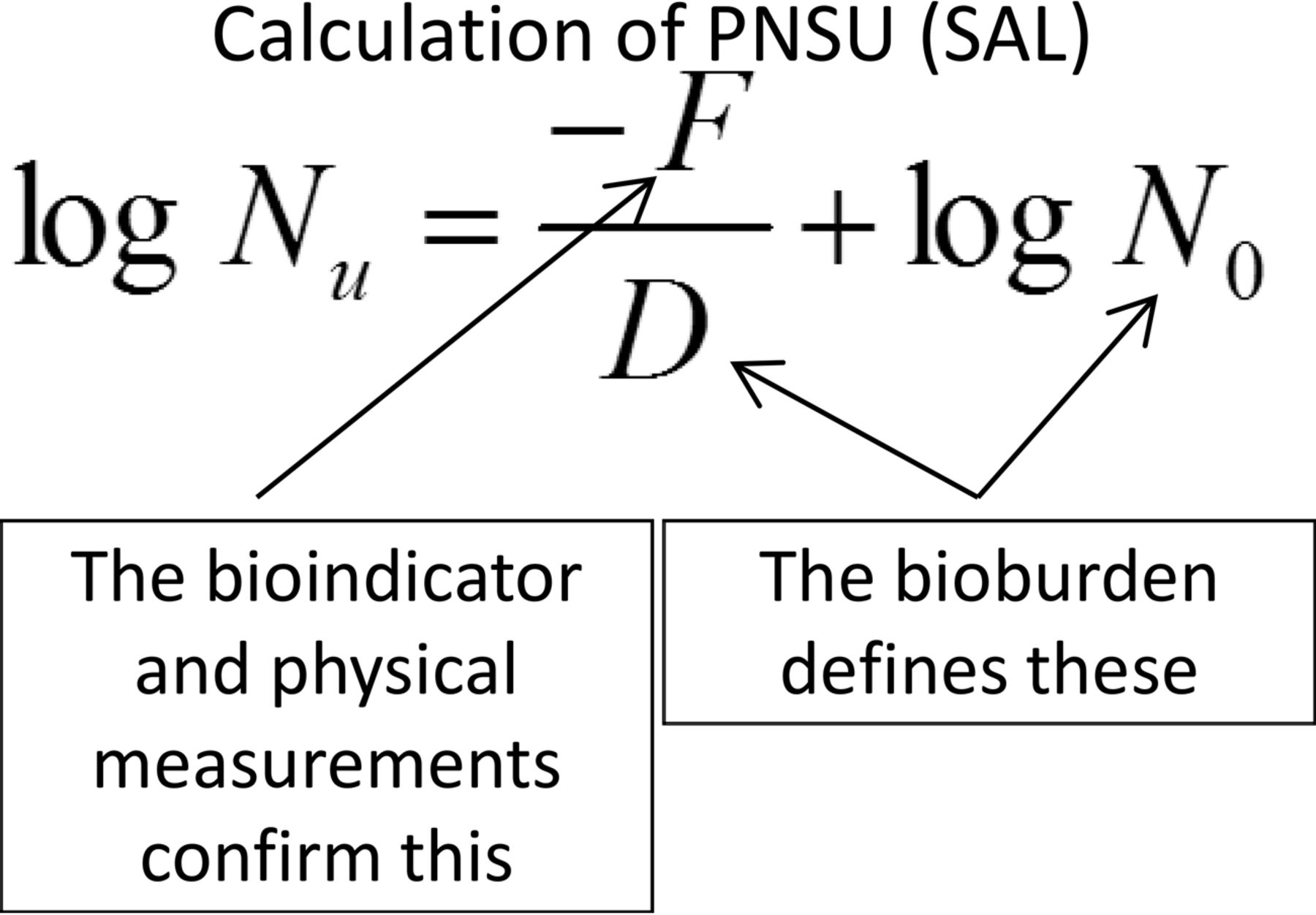
Calculation of probability of a non-sterile unit, where: Nu = PNSU; D = D-value of the natural bioburden (calculated or assumed); F = F-value (lethality) of the process; N0 = bioburden population.
Understanding that bioburden resistance and population are central to delivering safe drug products provides the means to close the process gap. A sterilization process must be able to consistently destroy the bioburden present with an appropriate margin of safety. Using a BI that is overly resistant, or process conditions based upon destruction of the BI, severely restricts the use of terminal processes. Of course the use of less aggressive sterilization conditions requires increased diligence on the part of the manufacturer to control bioburden population and resistance. The filling technologies available today make this a relatively easy task, as substantial human interaction with filling is less prevalent than in prior years.
Filling the Process Gap
The means to fill the process gap are already widely available. There are numerous sterile injectables available globally that are terminally sterilized below 121 °C, and some as low as 105 °C. Few, if any, of these terminal processes utilize Geobacillus stearothermophilus as the BI, as its thermal resistance is excessive in nearly all TS applications. G. stearothermophilus is a thermophilic spore-former that grows best at elevated temperatures (>50 °C), and it would be extremely unlikely for it to be present in ambient temperature processing environments. Spores of the genera Clostridia or Bacilli are common BI organisms used in these lower-temperature processes. There are strains in these genera with heat resistance that could prove useful in terminal sterilization processes in the 80–110 °C range. (This effort has focused on terminal sterilization by moist heat. An analogous argument can be made for terminal radiation of filled containers. That is still a rather uncommon process, but application of the same approach outlined for moist heat should be applicable.)
The filling of containers for these low-temperature TS processes should be aseptic to ensure the maximum control over the bioburden population. This would be unchanged from current practice, and some of the criticality of those filling operations would be reduced because viable monitoring of modern day aseptic filling largely recovers human-related microbes that are readily destroyed at temperatures at the expected sterilizing temperatures. (The costs of autoclave purchase and validation would be offset by the reduction in costs for investigations into environmental monitoring excursions and the potential for loss of an entire batch due to concerns over environmental conditions during its aseptic filling.)
For those readers that would dismiss low-temperature sterilization as an impractical and ineffective process, an accurate representation of its lethality against microorganisms of concern from a patient safety perspective is in order. The microorganisms in Table I are associated with a variety of human disease conditions, and availability of their moist heat resistance was necessary for inclusion in the table and calculations. Using the equation shown in Figure 2 for both an 80 °C process with a 20 min dwell period (F = 20) and 100 °C process with a 5 min dwell period (F = 5), the PNSU for several pathogenic microbes is presented in Table I. (The F = 20 and F = 5 values should be inserted into equation shown in Figure 2 to determine the estimated PNSU for each process.)
Calculated PNSU for Selected Pathogens at 80 °C/20 min & 100 °C/5 min*
The PNSU values in Table I are multiple orders of magnitude lower (better or safer) than the expected minimum of 1 × 10−6. Any suggestion that these processes are not sterilization processes is incorrect when it is acknowledged that the intent of the sterilization process is to destroy the pre-sterilization bioburden. As these processes are intended for use after aseptic filling with all of the associated controls utilized for that critical process, the presence of spores within the containers is unlikely. That these processes are not effective against all spore formers that might enter the fluid/container post-filtration and prior to sealing is irrelevant, as there is no ready means to prevent that eventuality where aseptic processing is used without a subsequent terminal sterilization.
Conclusion
It's past time to close the process gap that exists. The means to do so has been available for many years. Understanding the real target of the terminal sterilization process makes all the difference and allows for the implementation of a safer process for many current and future aseptically filled products. To ensure that patient-centered decisions rather than arbitrary specification setting based on misapprehension guides the process, scientific and engineering process experts must be involved. This must include those who are trained in clinical microbiology and infectious disease. The potential for a continuum of processes for sterile products exists (see Figure 3).
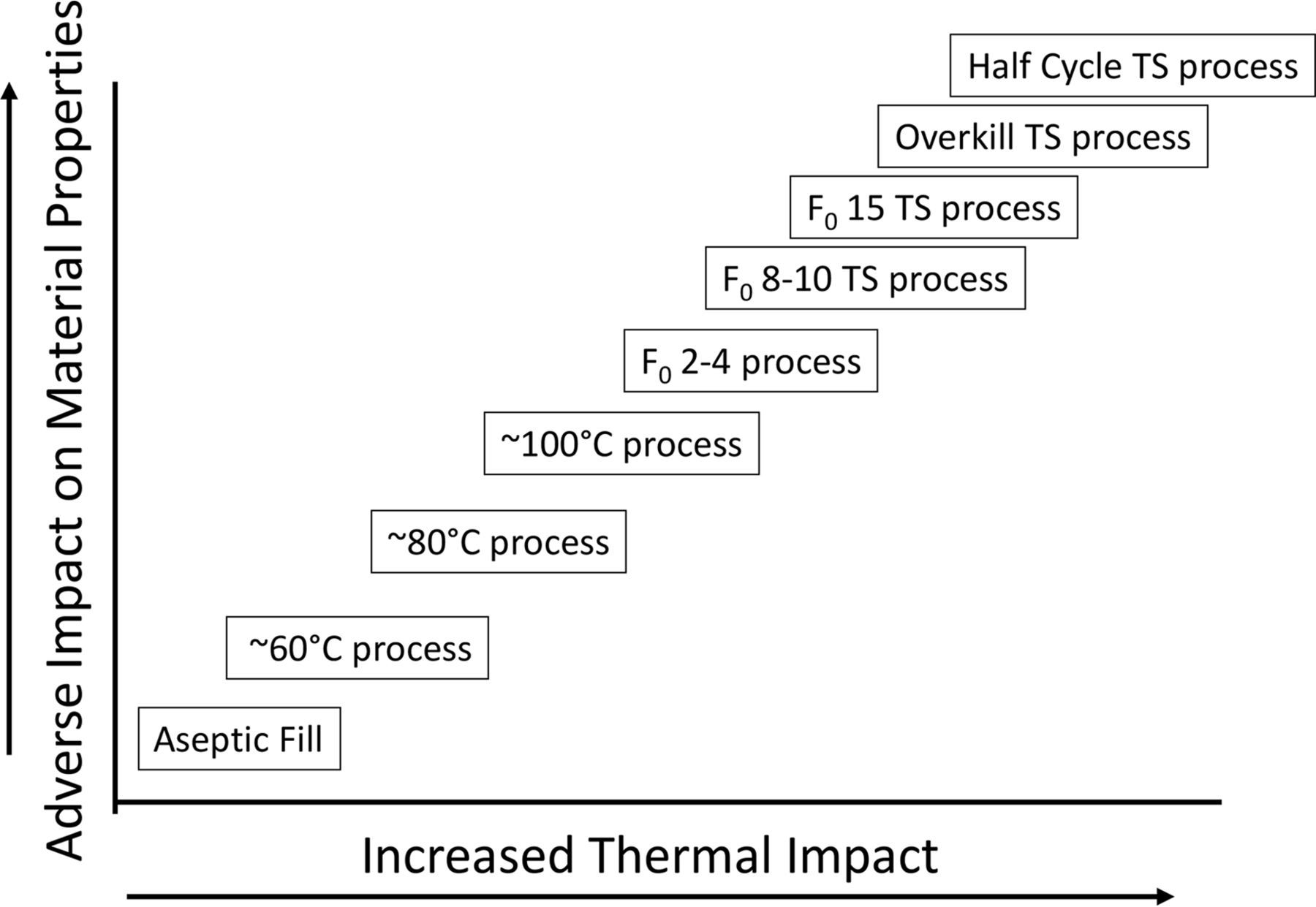
Terminal sterilization processing continuum.
Producing more sterile products with a terminal sterilization step subsequent to an aseptic fill offers a myriad of benefits. The goal in the production of sterile drugs should always be to assure patient access to safe and efficacious products. Closing the process gap by using a combination of aseptic processing and subsequent terminal sterilization ensures everyone wins:
Patients—receive safer and more stable product with fewer degradation materials; greater availability of drugs, increased safety.
Regulators—see above and below.
Manufacturers—easier aseptic processing operations, fewer expensive and fruitless environmental monitoring activities, reduced inconclusive environmental excursion reviews, greater reliability of supply, overcoming some of the deficiencies associated with aging facilities, reduced reliance on personnel performance, and reduced reliance on sterility testing.
Conflict of Interest Declaration
The author declares that he has no competing interests.
Acknowledgements
The assistance of Dr. James Akers and Mr. Russell Madsen in the preparation of this manuscript is greatly appreciated.
- © PDA, Inc. 2017




{kind=link}
{kind=link}
{kind=link}