
Abstract
Lyophilization is commonly used to extend the shelf life of pharmaceutical products that are otherwise unstable when stored as a liquid formulation. However, the ability of a lyophilized drug, or other solid medium, to leach or extract substances from a pharmaceutical packaging material is not well characterized. To provide insight into this area of uncertainty, the extraction properties of a lyophilized drug product, the lyophilized drug product reconstituted in water, and several other solid and liquid media of varying polarity were determined using a glass vial with a butyl rubber stopper as a representative pharmaceutical packaging system. The results obtained in this study show that the extracting power of a medium, whether solid or liquid, was primarily a function of polarity. Thus, the amount of each extractable observed for the lyophilized and reconstituted drug product were in trend with the other solid and liquid media, respectively. Nevertheless, it was notable that the lyophilized drug product was able to leach substances from the stopper in quantifiable amounts, whereas the reconstituted drug product contained no detectable leachables. Using a mathematical relationship, it was determined that the extraction power of the lyophilized drug product was equivalent to a 50/50 isopropanol/water solution.
LAY ABSTRACT: Freeze drying is commonly used to extend the shelf life of pharmaceutical products that are otherwise unstable when stored as a liquid formulation. However, the propensity for substances to migrate from a pharmaceutical packaging material and into a solid drug formulation is not well characterized. To provide insight into this area of uncertainty, the migration of substances from a glass vial with a butyl rubber stopper and into a lyophilized drug product, the drug product reconstituted with water, as well as several solid and liquid media of varying polarity were assessed. The results obtained in this study show that the extracting power of a medium, whether solid or liquid, was primarily a function of polarity and thus could be related to one another. Furthermore, the results for the freeze-dried and reconstituted drug products were in trend with the other solid and liquid media tested, respectively, and showed that the freeze-dried drug was able to leach substances from the stopper in measureable amounts, whereas the reconstituted drug product contained no substances that had originated from the stopper.
1. Introduction
Lyophilization (also known as freeze drying) is a process that employs reduced temperature and pressure in place of heat to remove water from a substance. In the pharmaceutical industry, lyophilization is used as a non-destructive means of reducing a liquid formulation to its solid constituents. This process is primarily performed to increase the stability and shelf life of drug products that are otherwise chemically and/or biologically unstable in solution. Preservation via lyophilization is common for biological-type pharmaceuticals, which have the greatest difficulty achieving reasonable shelf lives in a liquid formulation. Consequently, as the development of these types of products increase, so too does the use of lyophilization in the pharmaceutical industry (1, 2).
As with any pharmaceutical product, it is necessary to establish that the packaging system used to store a lyophilized formulation is not introducing impurities that may negatively affect its safety and/or efficacy. This is typically achieved by performing extraction studies, whose purpose is to generate a profile of compounds that can be extracted from the material via various contact scenarios, and leachable studies, whose purpose is to determine if packaging-related substances have migrated into the product over its lifetime. The results obtained from an extraction study are typically used in the selection/characterization of a packaging configuration and/or its materials of construction, whereas the results of a leachable study serve to confirm the suitability of the packaging system during its use with the product.
The leaching of substances from a packaging system and into a solution is common and generally well characterized (3⇓⇓⇓–7). However, the interaction of a lyophilized product, or other solid medium, with a packaging system has not been thoroughly explored. Therefore, there is uncertainty as to what differences, if any, are present in the rate and/or magnitude for the leaching of substances from a packaging system by a lyophilized versus liquid drug formulation, or extracted by a solid versus liquid medium. Characterization of these interactions would be beneficial for the design of extractable studies for packaging systems used with lyophilized products, and would also provide a better understanding of the leaching characteristics associated with these formulations.
Accordingly, the goal of this study was to determine and compare the properties of solid and liquid media for the extraction/leaching of substances from the material(s) comprising a pharmaceutical packaging system. These properties were assessed using a glass vial and butyl rubber stopper as a representative pharmaceutical packaging system. A lyophilized drug product, the lyophilized drug product reconstituted into an aqueous solution, potassium chloride, activated charcoal, and isopropanol/water solutions of varying proportions were selected as representative solid and liquid media. Each medium was evaluated after contacting the packaging system for various durations in order to evaluate the rate of extraction occurring for each medium. To quantify the interaction, several cyclic hydrocarbon oligomers originating from the stopper material, which are known to be common extractables/leachables for similar packaging configurations, were targeted.
2. Experimental
2.1. Study Design
The experiments performed in this study were designed to evaluate the extraction/leaching properties of solid and liquid media for substances for a contacting material. The extraction properties of the media have a thermodynamic aspect (magnitude of extraction) and kinetic aspect (rate of extraction). In order to evaluate the magnitude of extraction, solid and liquid media representing both polar and non-polar properties were included. Polarity was selected as a relevant property for study because it has been demonstrated to be a valid descriptor of the affinity of one substance for another, colloquially described as “like dissolves like”. The kinetic aspect of extraction was evaluated by sampling each media after it had contact the vial/stopper system for various periods of time, thus allowing a trend evaluation to be performed for the mass accumulated versus time.
The compounds present in the packaging system that were targeted to quantify the extraction properties of the media were several polymerization termination by-products (oligomers) of the butyl rubber stopper material. Table I presents their structure and other relevant properties. Selection of these compounds as targets was justified based on their presence as leachables in lyophilized formulations stored in similar vial/stopper systems, which is a consequence of their volatility and ubiquitous presence in butyl rubber material. Commercially available reference material was not available for these compounds, nor is reference spectra in commercially available gas chromatography/mass spectrometry spectral libraries. However, the compounds have been characterized in the scientific literature (8, 9), including detailed information on their mass spectra, structure, and genesis in the polymerization process.
Information for Target Extractables. The exact structure for each of the brominated oligomer isomers is not known. Instead, a single set of values is presented for the structure listed as an approximation of their properties. Log Po/w and boiling point values were estimated using EPIWEB 4.1 software.
In addition to the extraction/migration experiment performed in this study, the total pool of each extractable compound in the stopper was determined. Here, the total pool is defined as the total amount of each compound present in the entire stopper. Establishing the total pool is necessary to prove that any trends observed in the results, such as the attainment of equilibrium, are not simply due to the depletion of the mass of the compound in the stopper. Additionally, the total pool is used for the calculation of the partition coefficient of each extractable.
A single ion monitoring gas chromatography/mass spectrometry (SIM-GC/MS)-based method was used to qualitatively and quantitatively analyze the oligomers targeted in this study. This methodology was selected primarily because these compounds were known to be suited to this technique, and also because it would provide the detection and quantification limits desired.
The amount of a substance extracted or leached by a medium in contact with a packaging system or material is a function of the affinity of the substance for that medium. In a biphasic system, such as that being evaluated in this study, this affinity is commonly represented by a partition coefficient (P) (7, 10–11). Accordingly, the partition coefficients for the target oligomers, when present at quantifiable concentrations, were calculated for further interpretation of the extraction properties of the media.
2.2. Materials and Reagents
All solvents/reagents were obtained from Burdick and Jackson (Morristown, NJ) or Sigma-Aldrich (St. Louis, MO). Water was produced in-house and had a resistivity of 18.2 MΩcm.
The identity of the manufacturing organization of the lyophilized drug product is not disclosed in order to protect its confidentiality. However, it is known that the vial contained 0.5 g of an antibiotic drug and that the packaging system consisted of a Bormioli 20 mL Type 1 glass vial and a Datwyler 20 mm bromobutyl rubber stopper. Additional unused vials and stoppers were obtained from the same organization for use in preparation of the other extraction samples so that all data obtained in this study were from the same packaging materials.
2.3. Preparation of Extraction Samples
The activated charcoal and potassium chloride media were washed prior to use. This was performed by placing an appropriate amount of each material into an empty solid phase extraction tube, attaching the tube to a vacuum manifold, and then rinsing with copious amounts of solvent. Specifically, water, methanol, and dichloromethane were used to rinse the activated charcoal, while only methanol and dichloromethane was used to rinse the potassium chloride due to its solubility in water. After washing, the material was thoroughly dried by flowing air over the material.
Isopropanol/water solutions were prepared volumetrically (v/v) with isopropanol proportions of 10%, 25%, 40%, 55%, and 70%. This was performed by adding the appropriate amount of isopropanol to an appropriately sized volumetric flask, bringing the flask to volume with water, and mixing the solution thoroughly. Each solution was stored in the tightly stoppered flask.
Multiple vials of each liquid or solid extraction medium were prepared to fulfill the needs of the study. Test samples for the solid media were prepared by weighing 0.5 g of the media into each vial. Test samples for the isopropanol/water solutions were prepared by adding 5.0 mL of solution to each vial. The vials were then sealed with a stopper and secured by an aluminum seal. The lyophilized drug product was used as is, whereas the reconstituted drug solution was prepared by adding 10.0 mL of purified water was added to each vial of drug product, which was then placed into an ultrasonic bath to aid dissolution. Once dissolved, the vial was re-sealed with its stopper and secured with an aluminum seal.
Extraction of the butyl rubber stoppers with each medium was facilitated by heating the test samples at 40 °C in an inverted position (stopper down, in contact with the media) for a time period of up to 16 weeks. Shorter time periods were used if equilibrium had been achieved, or if there was insufficient material to support the full 16 week study, which was the case for the lyophilized drug product. At each time period, three vials were removed and tested per medium.
2.4. Preparation of Total Pool Determination Samples
The total pool of each butyl rubber oligomer was determined using sequential extractions of the stopper material. To prepare each extract, a stopper was cut into quarters and placed into a boiling flask containing 100 mL of ethyl acetate. The solvent was then brought to, and maintained at, a reflux for about 2 h, after which it was cooled, decanted from the stopper pieces, and reserved for analysis. A fresh aliquot of ethyl acetate was added to the flask and the extraction was repeated in the same manner until the desired number of extractions had been performed.
3. Analytical Methodology
3.1. Preparation of Solid Media for Analysis via Solvent Extraction
The solid media were prepared for analysis via a solvent extraction technique. For each sample, 5.0 mL of extraction solvent was added directly to the sample vial. The extraction solvent consisted of ethyl acetate into which acetophenone-d5 was added at a concentration of 1 μg/mL for quantification purposes. Next, each vial was placed in an ultrasonic bath for a period of approximately 5 min to facilitate the extraction. Following sonication, the solution was transferred into a glass tube that was centrifuged for 2 min at 500 × g. The clear supernatant solution from the KCl sample was used directly for analysis. The supernatant solution from the activated charcoal sample was removed from the tube and filtered through a 0.45 μm polytetrafluoroethylene (PTFE) filter prior to analysis in order to ensure none of the charcoal material was present in the final analytical sample.
3.2. Preparation of Liquid Media for Analysis via Solid-phase Extraction (SPE)
SPE was employed to isolate the compounds of interest from each aqueous test solution followed by reconstitution in a concentrated fraction of gas chromatography (GC)-compatible solvent. A commercially obtained polystyrene divinyl benzene SPE column [Agilent (Santa Clara, CA), Mega Bond Elut Plexa, 6 mL, 500 mg] was used for the procedure. PTFE reservoirs were attached to the inlet side of the columns to increase capacity, if needed. A vacuum manifold was used to produce the desired flow of each solution through the stationary phase by generating negative pressure on the outlet side of the column. Each column was prepared for use by conditioning with 4 mL of ethyl acetate, 4 mL of methanol, and 4 mL of 15/85 isopropanol/water added in succession. This series of solvents was used to remove any trace-level impurities from the stationary phase or polymeric column material, purge the ethyl acetate from the stationary phase with a water-miscible solvent, and wet the stationary phase, respectively.
For each isopropanol/water sample, the contents of the vial were quantitatively transferred to the prepared column using 15/85 isopropanol/water. An appropriate volume of water was added to the column prior to the addition of sample to produce a final solution containing approximately 15% isopropanol in order to ensure adequate retention of the target compounds on the stationary phase. Following the addition of the sample, 200 μL of 10 μg/mL acetophenone-d5 in methanol was added for quantification purposes. The solution was mixed to ensure homogeneity and then pulled through the stationary phase at a rate of approximately 5 mL/min.
In order to be processed by SPE, the lyophilized drug was first reconstituted using 10 mL of 15/85 isopropanol/water. The reconstituted drug solution was then quantitatively transferred to a prepared SPE column using 15/85 isopropanol/water, followed by the addition of 200 μL of 10 μg/mL acetophenone-d5 in methanol for quantification purposes. The pH of this solution was adjusted to ≥9 using dilute sodium hydroxide (for example, 1 N) in order to ionize the drug matrix and minimize its retention on the SPE stationary phase. After mixing, the solution was pulled through the stationary phase at a rate of about 5 mL/min, and then 2 mL of a pH ≥9 15/85 isopropanol/water solution was pulled through the column as a rinse.
Once the stationary phase had been loaded with the isopropanol/water or drug product solution, maximum vacuum was applied to completely dry the stationary phase. Following the drying procedure, 2.8 mL of ethyl acetate was added to each column, pulled over the stationary phase at a rate of about 5 mL/min, and collected. Partial absorption of solvent by the stationary phase produced a final eluent volume of 2.0 mL with an acetophenone-d5 internal standard concertation of 1 μg/mL. The eluent was used directly for analysis.
3.3. SIM-GC/MS Analysis
3.3.1. Instrumental:
Quantification of each oligomer was performed using a SIM-GC/MS method. An Agilent 7890A gas chromatograph and an Agilent 5975C single quadrupole mass spectrometer were the specific instrument make and model comprising the GC/MS system. The operating parameters used for the method can be found in Table II.
SIM-GC/MS Method Operating Parameters
3.3.2. Quantification:
Reference standards were not commercially available for the extractable compounds targeted in this study, which precludes the use of such a standard to quantify of the amount of each compound present. Instead, acetophenone-d5 was used as a surrogate standard for quantification of the peak area response of each target compound obtained in the SIM-GC/MS analysis. Acetophenone-d5 was selected because it is known to chromatograph and recover well by GC and SPE, respectively. Although this compound did not provide an exact response match for each target compound, the purpose of the study is primarily for trend evaluation. Therefore, this approach was considered to be suitable.
The use of SIM without the availability of authentic reference material necessitated the use of a correction factor to normalize the single ion response, which is dependent on the fragmentation pattern of each compound. To calculate the correction factor, each target compound and the acetophenone-d5 standard were analyzed in full scan mode (29–700 daltons) and in single ion mode. The differences between these responses, and between the surrogate standard and target compounds, were then used to determine the appropriate correction factor.
3.4. Evaluation of Method Performance
Method performance was evaluated in two ways. First, the ability of the method to accurately and precisely recover the target compounds, which is primarily a function of the sample preparation procedure, was confirmed during development of the method. Second, the verification that the instrument was performing acceptably was performed for each analytical set.
Performance of the solvent extraction and SPE sample preparation procedure was evaluated via experiments that added known amount of the target compounds to the matrix, processed the matrix via the appropriate procedure, and assessed the amount that was able to be recovered. As a consequence of the absence of authentic standard material, a traditional spike and recovery experiment could not be performed. Thus, an alternate approach using the target compounds, which were obtained directly from the stoppers, was employed. In order to obtain specimens of each compound, the stopper was extracted with ethyl acetate facilitated by a soxhlet apparatus. This produced a concentrated solution that was spiked into the appropriate solid or liquid test matrix and then processed per the remaining procedural steps. The spiked samples were analyzed along with the original isopropanol or ethyl acetate extracts, and peak areas obtained from the samples were compared to the peak areas of the original extracts, accounting for any dilution, and the percent of each compound recovered was determined. Adequate recovery (85–105%) and repeatability results were obtained for all solutions and solid media with the exception of the activated charcoal material. The charcoal results were reproducible, but the recovery values were approximately 50–70% of the original solution. This is suspected to be a result of the binding power of this sorbent for the hydrophobic compounds being studied. Because the results were reproducible, a correction factor was applied during quantification in order to prevent any underestimation of the reported values.
Instrument performance was verified for each analytical set by assessing non-interference, sensitivity, linearity, and precision. Non-interference was established by analyzing an ethyl acetate solvent control to demonstrate the solvent used in the procedure, or the instrument background, was not introducing interfering responses in the chromatographic data. Sensitivity was assessed by injecting a 10 ng/mL acetophenone-d5 standard and verifying that it produced a signal-to-noise ratio of ≥10. Linearity of response was assessed by analyzing five acetophenone-d5 standards prepared over a range of 10 ng/mL to 10 μg/mL and verifying that the correlation coefficient of the best line trend was ≥0.995. Precision was assessed by making multiple injections of a 1 μg/mL acetophenoen-d5 standard and verifying that the percent relative standard deviation of the response was ≤10.
3.5. Controls
Controls were prepared for the solvent extraction and SPE sample preparation procedures by processing the extraction media that had not contacted the stopper. In the case of the charcoal and potassium chloride sorbents, each was extracted with ethyl acetate per the solvent extraction procedure. Similarly, the isopropanol/water solutions were prepared by SPE. All controls were then analyzed by the SIM-GC/MS method to demonstrate the sample preparation procedures were not artificially introducing the butyl rubber oligomers being tested.
3.6. Calculation of Partition Coefficients
In its simplest form, the partition coefficient is an expression of the ratio of the oligomer concentration in the stopper phase, referred to as the donor phase (CD), versus that present in the solid or liquid medium extraction medium, referred to as the receiving phase (CR), and takes the form of eq 1:
 In order to calculate the partition coefficient, eq 1 must be further extrapolated in order to allow the input of values which can be measured experimentally. Thus, the concentration in each phase can be reduced to its constituent variables to produce eq 2, where m is the total mass of the extractable in the donor (D) and receiving (R) phases and M is the mass of each phase:
In order to calculate the partition coefficient, eq 1 must be further extrapolated in order to allow the input of values which can be measured experimentally. Thus, the concentration in each phase can be reduced to its constituent variables to produce eq 2, where m is the total mass of the extractable in the donor (D) and receiving (R) phases and M is the mass of each phase:
 The mass of the donor phase (MD) is the mass of the stopper, which was determined to be 1.76 g with a standard deviation of 0.01 g (n = 6), and the mass of the receiving phase (MR) is the mass of each medium weighed at time of preparation, or the volume delivered converted to mass using the solvent's density. Using these values, P can be calculated using eq 3, in which the total mass of the extractable in the stopper (mD) is further defined as the total mass intrinsically present in the stopper, which is the experimentally determined total pool (T) minus that which was measured to have migrated into the medium:
The mass of the donor phase (MD) is the mass of the stopper, which was determined to be 1.76 g with a standard deviation of 0.01 g (n = 6), and the mass of the receiving phase (MR) is the mass of each medium weighed at time of preparation, or the volume delivered converted to mass using the solvent's density. Using these values, P can be calculated using eq 3, in which the total mass of the extractable in the stopper (mD) is further defined as the total mass intrinsically present in the stopper, which is the experimentally determined total pool (T) minus that which was measured to have migrated into the medium:
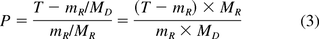 Because the partition coefficient can span several orders of magnitude, these values are expressed as the log10 in this study (Log P) for presentation and interpretation purposes.
Because the partition coefficient can span several orders of magnitude, these values are expressed as the log10 in this study (Log P) for presentation and interpretation purposes.
4. Results
4.1. Total Pool Determination
Table III contains the total pool measured for each target extractable from the six sequential extracts of the stopper, and Figure 1 presents a plot of the total amount of each compound extracted versus the total number of extracts performed. As can be seen in this figure, the trend in the total mass of each compound extracted has reached a plateau that is indicative of an exhaustive extraction being achieved. Furthermore, the mass extracted in the last preparation was <10% of the mass extracted in the first preparation, which is also consistent with the attainment of an exhaustive extraction as defined in International Organization for Standardization international standard 10993 (12).

Total pool determination. Plot of the total amount of each compound extracted versus the number of extractions shows the total extracted had reached asymptotic levels indicative of an exhaustive extraction. Values represent the mean of three replicate preparations.
Total Pool Amounts Measured for Each Target Extractable from Six Successive Extractions of a Single Stopper. Mean and standard deviation values were obtained from three replicate preparations. For comparison purposes, theoretical exhaustively extracted μg/g values are calculated for each medium. These represent the value that would be achieved if the total pool of each oligomer had been completely extracted into the media.
4.2. Lyophilized Drug Product, Reconstituted Drug Product, and Solid Extraction Media
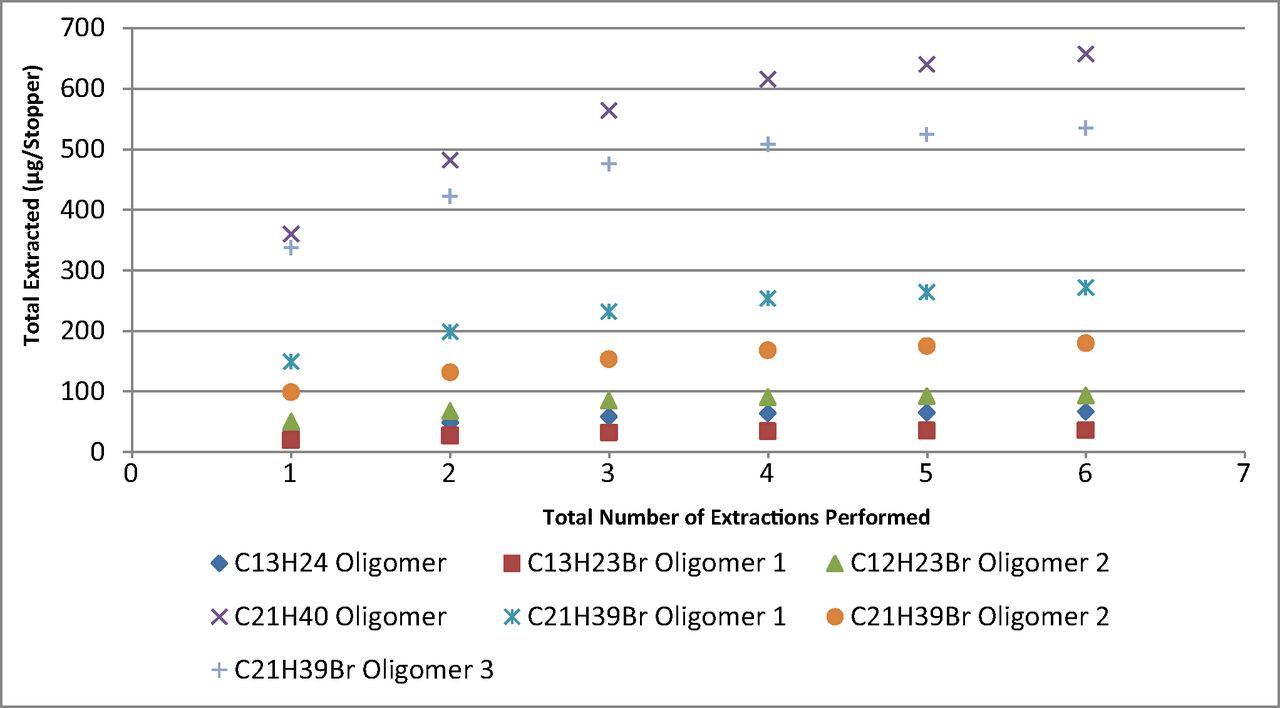
The trend in the total extracted amount of each rubber oligomer versus time for the lyophilized drug product and activated charcoal, as can be seen in Figure 2, shows differences in the rate (mass/time) of extraction. Additionally, it is evident that some of the oligomers have reached, or are approaching, equilibrium at the final time point analyzed (12 weeks) for the lyophilized drug product, whereas the activated charcoal results showed a relatively linear increase in the extracted amount of each oligomer up to the final time point tested (16 weeks). Although the potassium chloride medium did not produce quantifiable data, the trends in the responses did not change significantly from the first time point tested, suggesting equilibrium had been attained.

Plot of the amount extracted versus time for the lyophilized drug product and activated charcoal from Week 2 through the final time point analyzed. Error bars represent 1 standard deviation from the mean.
For each medium, a significant variation in the amount of each oligomer extracted and the calculated Log P was observed at the final time point analyzed (Table IV). No detectable response was present for any of the target oligomers in the reconstituted drug product.
Concentration and Log P of Each Oligomer in the Lyophilized Drug Product, the Reconstituted Drug Product Solution, Potassium Chloride, and Activated Charcoal at the Final Time Point. Mean (x̄) and standard deviation (σ) values were obtained from three replicate preparations. <QL (quantification limit): Compound was detected, but the response was below the method quantification limit of 0.04 μg/g for the lyophilized drug product, 0.002 μg/mL for the reconstituted drug product, and 0.1 μg/g for potassium chloride and activated charcoal. Log P values preceded by < indicate equilibrium was not achieved and thus final value may be less than reported. Log P values preceded by ≥ indicate value was less than the QL, and thus the QL concentration was used to approximate the value which may be greater than that reported. ND: Not detected; NA: Not applicable; -: No quantitative data available for calculation of Log P.
4.3. Isopropanol/Water Solutions
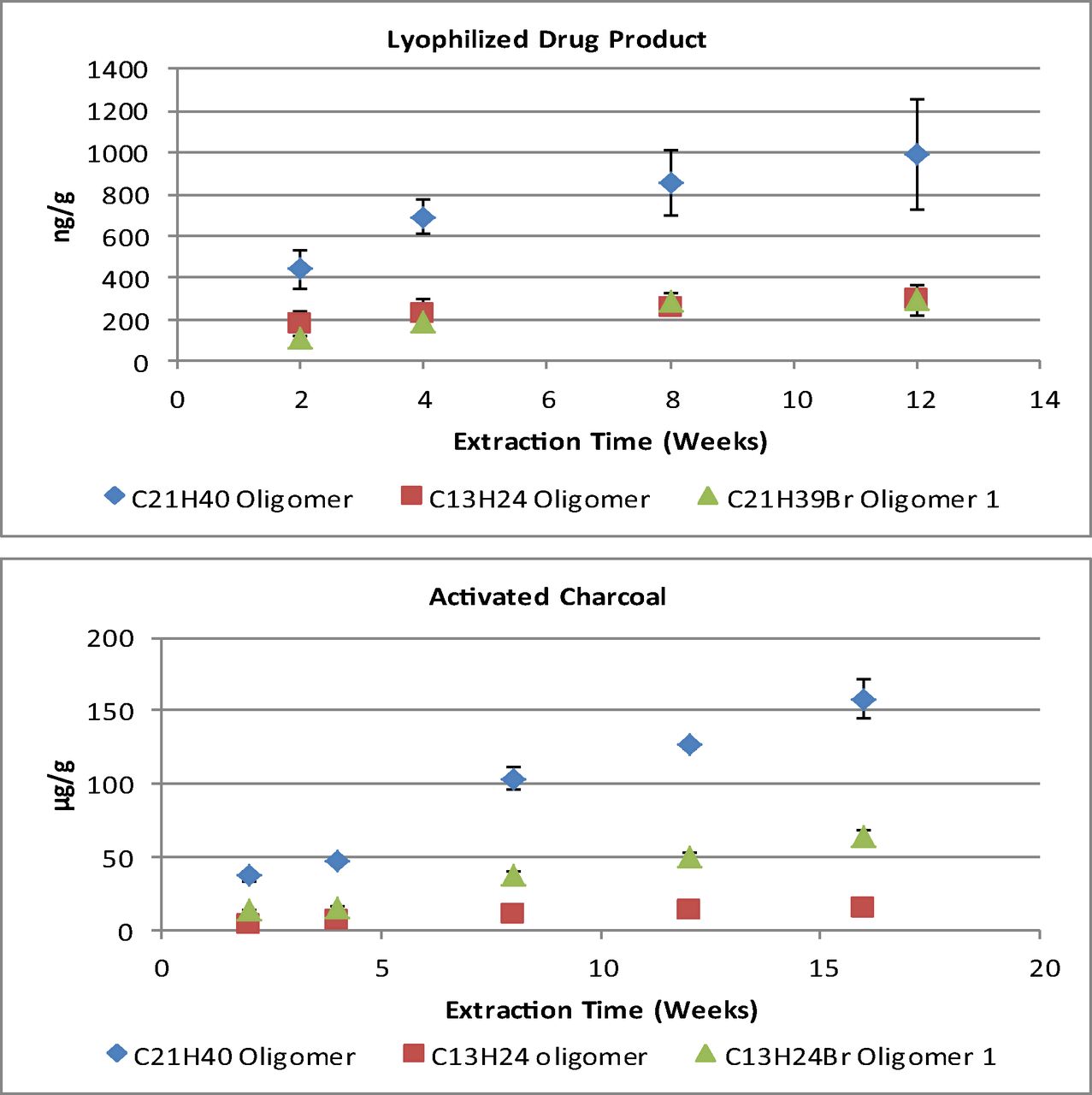
As with the solid media, the rate of extraction of the rubber oligomers, and the timed needed to reach equilibrium, differ in each of the liquid media tested. This is illustrated in Figure 3, which presents a plot of the quantifiable amounts of the C13H24 and C21H40 oligomers extracted in the 70%, 55%, and 40% isopropanol/water solutions over the course of the study. The 10% and 25% isopropanol/water solutions had no detectable or quantifiable responses, respectively. Instead, a review of the peak area response was performed for the oligomers detected in the 25% isopropanol solution and showed that an apparent equilibrium had been achieved at the first time point analyzed.

Plot of the total extracted amount (μg/g extraction solvent) of the C13H24 and C21H40 oligomers extracted in quantifiable amounts for the isopropanol/water extraction solvents versus the duration of the extraction. Error bars reflect 1 standard deviation from the mean (n = 3).
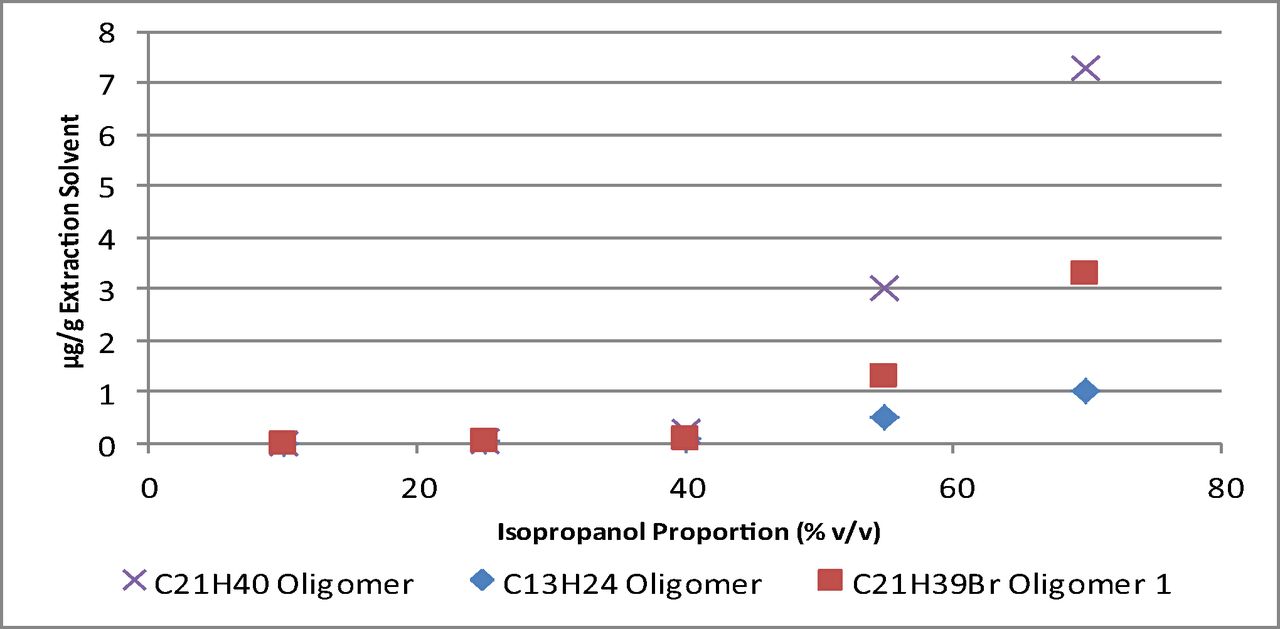
A summary of the amount of each oligomer extracted, and its calculated Log P, at the final time point analyzed for each isopropanol/water solution can be found in Table V. Here, it is apparent that an increase in isopropanol proportion corresponds to a general increase in the amount extracted. However, when viewing a plot of the amount of each compound extracted versus the proportion of isopropanol, as in Figure 4, it is apparent that this increase follows a non-linear trend.

Plot of the amount of the C13H24, C21H40, and C21H39Br Oligomer 1 extracted at the final time point versus the isopropanol proportion of the extraction solvent.
Concentration and Log P of Each Oligomer Determined at the Last Time Point for the Isopropanol/Water Solutions. Mean and standard deviation values were obtained from three replicate preparations. Log P values preceded by ≤ indicate equilibrium may not have been achieved and thus the final value may be less than that reported. Log P values preceded by ≥ indicate value was less than the QL, and thus the QL concentration was used to approximate the value which may be greater than that reported. ND: Compound was not detected; NA: Not applicable. <QL: Compound was detected, but the response was below the method quantification limit of approximately 0.01 μg/g.
5. Discussion
As a first step toward achieving the goal of this study, an assessment of the extraction power of each solid and liquid medium is necessary. Using the partition coefficients determined in this study, it is clear that the polarity of the matrix, whether solid or liquid, is the predominant characteristic affecting the magnitude of oligomers extracted from the stopper. For example, the lowest Log P values are observed for the activated charcoal medium and the solutions with the highest proportion of isopropanol, which represent the least polar solid and liquid media, respectively. Similarly, the highest Log P values, or the absence of detectable levels of extractables/leachables, were observed for the most polar media, which were potassium chloride, solutions with the lowest isopropanol proportion, and the reconstituted drug product. Considering the fact that the extractables targeted in this study are all hydrophobic (Log P ≥6), and thus their affinity for more polar media is minimal, the observation that the non-polar media possess a higher extraction power is logical.
In addition to the extraction power of the media, an evaluation of the kinetic properties of the extraction is necessary to fulfill the goal of this study. To this end, there are two relevant observations that can be made based on the results obtained in this study. First, the magnitude of the rate of migration (mass/unit time) corresponds to the maximum amount of each oligomer that can be accumulated in each medium. Second, the amount of time necessary to reach an apparent equilibrium also corresponds to the total mass extracted, with the media having a lower extraction power, such as the 40% isopropanol solution or potassium chloride, reaching an apparent equilibrium more quickly than those with greater extraction power. The first observation is consistent with the principles of diffusion theory, namely the magnitude of the mass/unit time depending on the concentration gradient between the stopper phase and the contacting medium. The second observation suggests that the attainment of equilibrium is at least partially related to the magnitude of the accumulation end point in the contacting medium. For example, when other factors are equal, the media with lower extraction power reach equilibrium more quickly due to the fact that less mass is required to achieve a state of equilibrium.
Beyond these observations, it was not possible to further determine whether the kinetic properties of media with the same state (solid or liquid) but different extraction power, or media of different state but similar extraction power, have meaningful differences in the kinetics of the extraction process. This is a consequence of the complexity of the interaction occurring in each media, the relatively small data set available for interpretation, as well as other factors such as the potential for the rate of migration of the oligomers through the rubber material to become rate-determining.
Because polarity was shown to be a valid descriptor of extraction power regardless of whether the extracting medium is a liquid or solid, a relationship can be established linking the extraction power of media in different states. To illustrate this, a relationship can be established for the isopropanol/water solutions and lyophilized drug product by first plotting the Log P values obtained for each oligomer (Table V) versus the proportion of isopropanol in the solution and obtaining equations for the best-fit trend line. Once established, the Log P of each oligomer quantified in the lyophilized drug product (Table IV) was entered into the corresponding equation and the corresponding isopropanol/water proportion was calculated. Table VI contains the best-fit equation determined for each oligomer, the coefficient of determination (r2) of the best-fit trend line, and the isopropanol/water proportion calculated based on the Log P values from the lyophilized drug product. With the exception of the C12H23Br Oligomer 2, this relationship determined that the oligomers would require an isopropanol/water proportion of approximately 50% in order to mimic the extracting power of the lyophilized drug product.
Equations Generated from the Best Fit Trend Lines Obtained from a Plot of the Isopropanol/Water Proportion Versus the Resulting Log P Values from Table V for Each Extractable Obtained for the 25%, 40%, 55%, and 70% Isopropanol Extraction Media. By adding the Log P values for the quantifiable extractables from the lyophilized drug product from Table IV, the percentage of isopropanol in the extraction solvent required to mimic the extraction power of the lyophilized drug product is determined.
The results obtained for the lyophilized and reconstituted drug product, while contrasting one another, are consistent with the results obtained for the other solid and liquid media, respectively. For example, the level of extractables observed in the lyophilized drug product is greater than that observed in potassium chloride but much less than what was observed in the activated charcoal. This is consistent with the drug possessing a polarity modestly less than potassium chloride, which is a polar salt, but much greater than activated charcoal, which is a non-polar substance. Similarly, the fact that no extractables were observed for the reconstituted drug product is consistent with 10% isopropanol/water, which is the other medium with the highest water proportion tested. Thus, the lack of any detectable oligomers in these media can be attributed to the fact that both are highly aqueous solutions.
As previously noted, the results obtained for the isopropanol/water solution extracts showed that the increase in amount extracted versus the isopropanol proportion of the solution follows a non-linear trend (Figure 4). However, a similar trend was reported in a previously published study (13) for dioctyl phthalate extracted from polyvinyl chloride material by ethanol/water solutions. This provides evidence that the trend observed in this study is a function of the solubilization properties of this solvent system rather than something more specific to the packaging system and/or compounds tested. Additionally, the authors proposed that differences in structuring of the ethanol molecules at different proportions of ethanol/water may be responsible for the non-linear nature of this trend.
6. Conclusion
In general, the extracting power of each solid and liquid medium was observed to be a function of polarity. As a result, it was demonstrated that the extraction power of a solid and liquid medium could be mathematically related.
Considering the kinetics of extraction, it was observed that the magnitude of the observed rate (mass/unit time) is a function of the concentration gradient, with those media hosting the highest extractable amount having the highest mass/unit time values. Similarly, longer contact durations were required to reach equilibrium for the media with the higher extraction power, which is indicative of the mass movement required to achieve equilibrium being a limiting factor when other variables are equal.
The results for the lyophilized and reconstituted drug product were consistent and in trend with the other solid and liquid media. Nonetheless, the discrepancy between the lyophilized versus reconstituted forms illustrates the potential for leaching to occur for a lyophilized drug product versus one stored as a dilute aqueous solution. Furthermore, the mathematical relationship established in this study demonstrated that the extraction power of the lyophilized product was comparable to a 50% isopropanol solution.
Conflict of Interest Declaration
The author declares he has no competing interests.
Acknowledgments
The author would like to thank Julie Sutcliffe for assisting with the review and formatting of this paper.
- © PDA, Inc. 2017




{kind=link}
{kind=link}
{kind=link}
{kind=link}