
Abstract
During the processes involved in pharmaceutical manufacturing, particulate matter may be introduced into a product from a variety of sources and at different points in the manufacturing process. Companies design quality at the beginning of the process to ensure against defects and strive to manufacture products that meet the pharmacopeial standard of being “practically/essentially free” of particles, which can be challenging, though necessary. As particulate matter recalls are predominantly associated with parenteral products, most companies employ a quality risk management program to identify critical parameters or conditions that could affect product quality or patient safety and incorporate systemic and procedural controls to mitigate or reduce the probability of their occurrence. Yet, determining where particulates are most likely to enter the process, what types of materials are most vulnerable, and how the size and number of particles might affect product quality can be very complex. Visual inspection and sampling of the manufactured drug product are designed to control the risk of particulate contamination; building prevention controls will ensure sustainability. This concept paper highlights the necessity of a more thorough understanding of the failure mechanisms that result in particle contamination across a range of products, such as elastomeric components and glass, and processes, such as the formulation and filling of injectables. The goal is to identify process steps within the end-to-end manufacturing process that are most critical to particle generation and entering of visible particles into the final drug product.
LAY ABSTRACT: This concept paper highlights the necessity of a more thorough understanding of the failure mechanisms that result in particle contamination across a range of products, such as elastomeric components and glass, and processes, such as the formulation and filling of injectables. The goal is to identify process steps within the end-to-end manufacturing process that are most critical to particle generation and entering of visible particles into the final drug product.
1. Introduction
During the processes involved in pharmaceutical manufacturing, particulate matter may be introduced into a product from a variety of sources and at different points in the manufacturing process. Companies design quality at the beginning of the process to ensure against defects and strive to manufacture products that meet the pharmacopeial standard of being “practically/essentially free” of particles, which can be challenging, though necessary (1). As particulate matter recalls are predominantly associated with parenteral products (2), most companies employ a quality risk management program to identify critical parameters or conditions that could affect product quality or patient safety and incorporate systemic and procedural controls to mitigate or reduce the probability of their occurrence (3). Yet, determining where particulates are most likely to enter the process, what types of materials are most vulnerable, and how the size and number of particles might affect product quality can be very complex (4). Visual inspection and sampling of the manufactured drug product are designed to control the risk of particulate contamination; building prevention controls will ensure sustainability.
This concept paper highlights the necessity of a more thorough understanding of the failure mechanisms that result in particle contamination across a range of products, such as elastomeric components and glass, and processes, such as the formulation and filling of injectables. The goal is to identify process steps within the end-to-end manufacturing process that are most critical to particle generation and entering of visible particles into the final drug product. A failure mode effects analysis (FMEA) was selected as a potential approach that allows for an appropriate amount of flexibility so that it can be used to produce a high-level assessment of an entire process as well as focusing on a single operation to identify where and how it may fail. FMEA has proven effective for evaluating equipment and systems failure modes and can be applied to hardware, software, and procedures. The approach allows identification of component failures as well as their causes and effects on the system, providing needed information to develop monitoring programs for key areas and prioritizing the level of risk. The emphasis in conducting an FMEA is prevention, learning where the risks of failure are to correct the process and avert the possible risk of harm to the patient.
The analyses conducted and reported in this points to consider document evaluated the risk of visible particle contamination for the supply of elastomers and glass, and for the formulation and filling of an injectable pharmaceutical product, thereby covering the end-to-end manufacturing process of parenteral drug applications.
2. Scope
The cross-functional teams comprised of glass suppliers, elastomer suppliers, and fill finish operations entities each completed an FMEA to ascertain the failure mechanisms that result in particle contamination for a range of components. Components that display the current state of materials used in industry as well as the best-in-class manufacturing processes were selected. Components were chosen for glass suppliers, elastomer suppliers, and formulation and filling manufacturers (indicated in the cross divisional team setup in Figure 1).

Workstream alignment.
In order to be broadly applicable and to capture the most representative failure modes, all applicable elastomeric components were assessed including ready-to-use (RTU) and ready-to-sterilize (RTS) material. A variety of glass components were included for the assessment including RTU syringes, bulk-filled vials, and bulk-filled molded vials or bottles. During selection of the processes for pharma product manufacturing, the materials from the supplier FMEAs were used as the basis for the selection. This selection set the basis for the selection of manufacturing processes at the pharma companies, which were chosen to display a range of formulation and filling processes. Prefilled syringes and lyophilized product were chosen based on their capacity to be representative of the sufficiently broad range of opportunities for loose particulate introduction into the final product.
3. Process
3.1. Elastomer Components
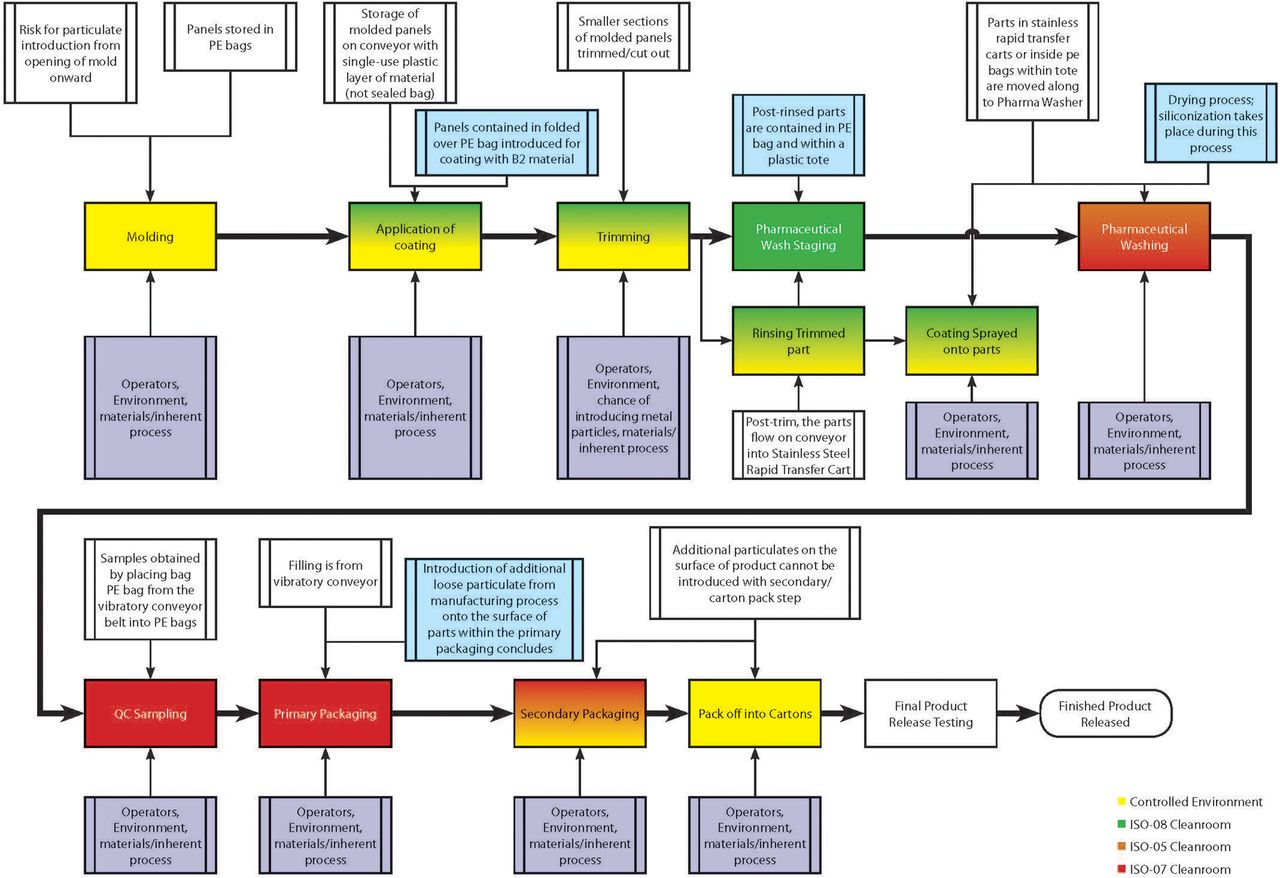
Rather than completing a detailed FMEA for bulk elastomeric products, the team narrowed the focus of the analysis to only RTS and RTU products. Prior to conducting the FMEAs for RTS and RTU product lines, all involved suppliers created high-level process maps for the analysis and then prepared a detailed process map for both RTS (Figure 2) and RTU (Figure 3) product types [and then reconciled areas of differentiation (Figure 4)].

RTS process map.

RTU process map.

High-level process map.
3.2. Glass Components
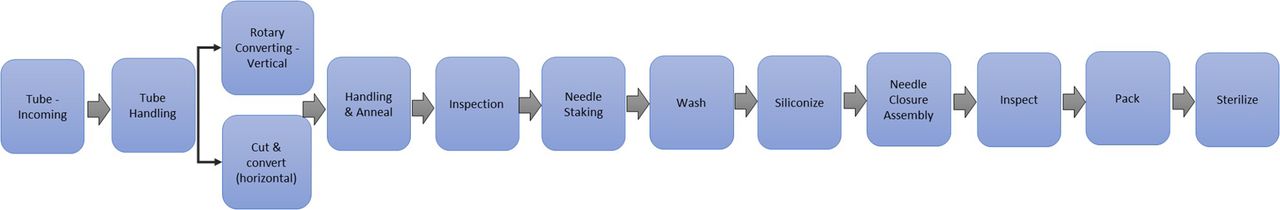
In order to be broadly applicable and capture the most representative failure modes, a variety of glass components were included in the assessment including RTU syringes, bulk-filled vials, and bulk-filled molded vials or bottles. Although not specifically included, the FMEA results for the RTU syringe are relevant to other RTU container formats. Prior to conducting the FMEAs for syringe, vial, and bulk bottle product lines, all suppliers created high-level process maps for the analysis (Figures 5⇓–7).
High-level process map bulk bottle.
High-level process map bulk vial.

High-level process map RTU syringe.
3.3. Fill and Finish of Injectable Pharmaceutical Product
Detailed process flow maps were constructed for two specific processes; solution-based API in a prefilled syringe and a lyophilized API in an elastomer-stoppered vial. The completed maps followed the product manufacturing process from formulation, filling, and through finishing of the final product in its primary packaging (filled syringe or capped & sealed vial).
Separate FMEAs were developed for each of these processes using the following process flow maps, taking into consideration the:
manufacturing environment;
manufacturing process and equipment;
entry of materials and components into the filling zone;
filling process, equipment, and environment;
personnel behaviors; and
miscellaneous commodities and support processes.
3.3.1. Prefilled Syringe with Solution Product:
The team prepared a process flow map for the manufacture of a simple solution product in a prefilled syringe format. The primary steps followed the manufacturing process from formulation of the product solution through sterile filtration into the sterile, product-holding vessel through filling into syringes to insertion of the plunger stopper (Figure 8).

Process map solution product in syringe.
Using this process flow map, the team prepared a detailed FMEA that looked at the potential entry points in the manufacturing process for product contamination by visible particulates (consolidated FMEA results will be published with the full technical report from this team; templates will be available for use). Each step in the process was identified and assessed, including the potential permutations associated with different levels of risk present considering the following factors:
Filling environment
Conventional filling line (Classical ISO 5)
RABS lines
Isolator lines
Commodity presentation
Syringe barrels
Bulk supply
Ready-to-use (RTU)
Plunger stoppers
Bulk supply
Ready-to-sterilize (RTS)
Ready-to-use (RTU)
3.3.2. Lyophilized Product in Vial:
The team prepared a process flow map for the manufacture of a lyophilized product in a glass vial with elastomer stopper format. This process flow map identified the primary steps of the manufacturing process from formulation of the product solution through sterile filtration into the sterile product–holding vessel through filling into vials, partial insertion of the stopper, lyophilization, full seating of the stopper, and capping (Figure 9).

Process map lyophilized product in stoppered vials.
Using this process flow map the team prepared a detailed FMEA that looked at the potential entry points in the manufacturing process for product contamination by visible particulates. Each step in the process was identified and assessed, including the potential permutations associated with different levels of risk present considering the following factors:
Filling environment
Conventional filling line (Classical ISO 5)
Restricted Access Barrier Systems (RABS) lines
Isolator lines
Commodity presentation
Bulk supplied vials
Depyrogenation/sterilization in oven
Depyrogenation/sterilization in tunnel
Stoppers
Bulk supply
Ready-to-sterilize (RTS)
Ready-to-use (RTU)
Lyophilizer
Partially stoppered vial transport cabinets
Simple transport cabinets
Self-contained HEPA transport cabinets
4. Risk Scoring and Levels
An FMEA considers three components of a problem—severity, frequency, and detection. This FMEA assessment used a scale of 1–5 for each of these three factors. For the frequency and detection scale, five levels were used in the assessment of each defined failure mode (see Table I).
Frequency and Detection Scale
Table II highlights the defined effect and detection that is correlated to an assigned numerical value.
Severity Scale
Based on their experience in the pharma industry, the participants selected a severity level of 3 (Level 3 = Moderate) for impact to the product and customer from the presence of any loose particulate. Each circumstance needs to be evaluated individually, which may require a health hazard assessment to better determine the severity. However, the presence of biological particulate was a bit higher at a severity level of 5 (Critical) because of the potential impact of rendering the packaged drug product adulterated.
4.1. Assumptions Regarding Frequency and Detection
Values assigned for the probability of observing a specific failure mode (frequency) were based on general industry experience focused on obtaining target areas for process improvement and reduction of particle contamination risk for later phases of the Task Force plan.
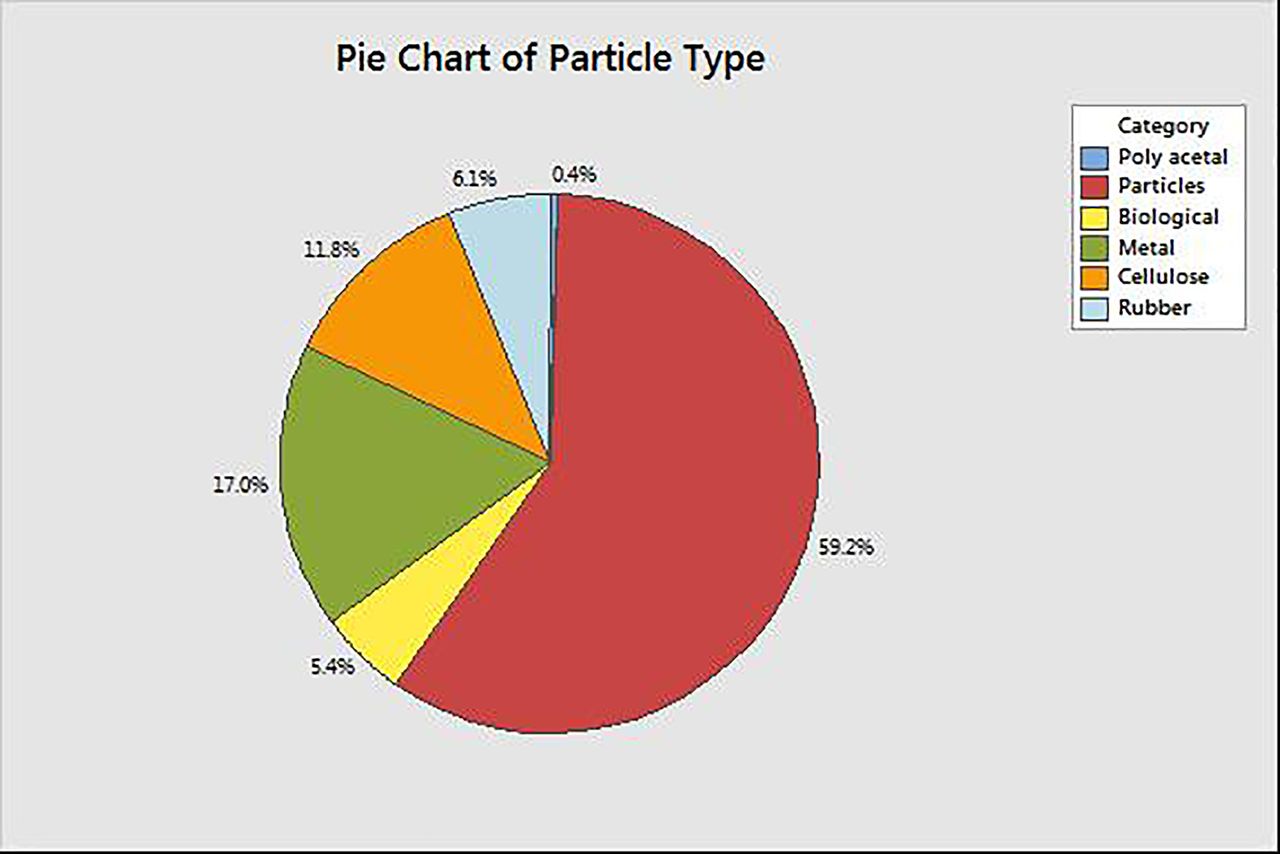
For the elastomer FMEA, customer complaints received from all three elastomeric manufacturers about loose particulate were evaluated (Figure 10). Allocating values to specific process points where particles were introduced, however, was not always feasible as the root cause for all complaints did not provide that level of specificity. To address the disconnect, a larger value for occurrence was assigned to process points of reduced existing controls.

Occurrence rate for specific particle types (observed based on customer complaints, PDA 2014 visual inspection survey).
The score for detection was derived based on the existing systems or methods used for the detection of loose particulate at specific process points or on the existing procedural controls applied at those same process points. The type of defect and its location within the container also contributed to the detection score. In the FMEAs, scoring accounted for detection of particles within containers and on elastomers that could be as small as 100 µm (4).
Within the glass team, the score for detection was difficult to score quantitatively because the wide variety of particle sizes, inspection methodologies, and systems. A general understanding among the glass manufacturers is that the type and location of the defect within the container would also contribute to the detection score. In the glass FMEA, scoring is based on detection of particles that can be as small as 100 µm in an empty glass container. Typical inspection methods are sensitive down to 200–300 µm, indicating that such methods are not sufficient for the reliable detection of particle sizes down to 100 µm. In general, glass particles can form in a range of sizes, with the smaller sizes being more difficult to inspect; for this reason, smaller glass particles were ranked as a 4 for the detection score. Fibrous particles were scored as a 2, as they tend to be larger in most cases. Whether particles are introduced before or after a 100% inspection was also considered in the scoring.
It is important to note that scoring depends on many factors, including the container geometry, manufacturing process, controls, operations, and environment. The scoring provided here is intended to be generally representative of typical operations; however, specific scoring would be different for each manufacturing operation and product. Where this FMEA is used as a starting template, existing prevention and detection controls should be used to score the specific severity, probability of occurrence, and detection for that instance.
5. Highlights and Opportunities for Improvement
FMEAs were prepared for the various manufacturing processes ranging from elastomer manufacturing, glass forming, and fill finish operations at pharma companies; some general topics were identified across the industry as particle entry routes throughout manufacturing (Table III). Collaborative efforts for information sharing were a common issue; however, a few common particle entry areas were identified. It was agreed that the most common were clean rooms, secondary packaging, and cutting processes. The involvement of secondary component manufacturers was deemed to be a necessity.
Summary of Opportunities for Improvement within Teams
5.1. Elastomer Component Process Improvements
A number of recommended best practices, improvements, and controls are listed for each of the failure modes in the FMEA. The first FMEA revision completed for the RTS process yielded 104 rows of failure modes with an assessment of the risk, the probability of occurrence, and controls/means of detection currently in place. Sorting of defined failure modes using a calculated risk priority number threshold of >30 yielded 14 specific failure modes within the molding through trimming process that have the potential of introducing metal particles, cellulose, biological contaminants, or other particulates. Once additional input is gathered from completion of actions by other subgroup workstreams, viable studies will be defined for the process steps within these areas that warrant further evaluation to determine feasible opportunities for process improvement.
The first FMEA revision completed for the RTU process yielded 117 rows of failure modes with an assessment of the risk, the probability of occurrence and controls/means of detection currently in place. Sorting of defined failure modes using a calculated risk priority number threshold of >30 yielded two specific failure modes within process steps associated with handling semifinished product (postmolding) that have the potential of introducing particulates. Once additional input is gathered from completion of actions by other subgroup workstreams, viable engineering studies will be defined for the process steps within these areas that warrant further evaluation to determine feasible opportunities for process improvement.
A number of controls, best practices, and best-in-class process methods were reviewed for each of the failure modes in the FMEA.
5.2. Glass Component Process Improvements
During the FMEA, the following process areas were identified with RPN values that indicated a high risk of particle introduction:
incoming glass particles on tubing;
forming steps where mechanical glass cutting is involved;
particles from the operative environment during RTU processing (wash, siliconize, needle closure); and
fibrous particles originating from RTU secondary components (tub, nest, Tyvek sheet, bags).
A number of best controls and best practices for process methods are listed for each of the failure modes contained in the FMEA. Several critical best practices are summarized here. Organizations should consider their internal processes and fully evaluate their specific control systems before implementation of new processes. Some considerations are noted below:
Minimize glass breakage by understanding and minimizing sources of glass surface damage and implementation of sensors to prevent overloading and/or misfeeds of glass components during handling.
Implement procedures or methods for detection, prevention, and reaction to broken tubes and vials throughput the process. Detection procedures could be automated. Reaction should isolate and minimize the impact of glass chips and particles from breakage.
Implement additional wash steps, properly performed, for tubing and containers to reduce contamination.
Implement additional equipment and/or environmental controls in the vicinity of systematic particle generation (i.e., glass-cutting steps, components, people, equipment) to prevent contamination of the container.
Minimize exposure of materials introduced in less restrictive areas to particles originating from packaging materials such as cardboard or other contaminants that may be present. Specify and properly introduce clean components (e.g., tubs, nests) into the cleanroom and verify cleanliness by measurement where possible. Clean and use proper pass-throughs for all equipment and materials being introduced into the cleanroom.
Minimize or eliminate operators from process areas. Isolate contaminations where operator activity is high.
Design equipment to avoid or isolate particle generation.
Adopt proper cleanroom maintenance and cleaning procedures with appropriate frequency, focusing on critical surfaces.
5.3. Formulation and Filling of an Injectable Pharmaceutical Product
During the FMEA, the following process areas were identified with RPN values that indicated high risk of particle introduction:
Filling area design
Conventional; RABS; Isolators
Integration of sterilizers, filling lines, and secondary processes (lyophilization and capping)
Lyophilizer
Filling line setup activities
Entry of commodities (containers and stoppers) and materials into the filling space
Interventions into the filling process—direct or indirect
Addition of stoppers to the stopper bowl during filling
Transfer of partially stoppered containers from the filling line to the lyophilizer
Preparation, storage, and handling of materials and commodities entering into the filling space
Personnel gowning and aseptic behaviors
A number of recommended best practices, improvements, and controls are listed for each of the failure modes in the FMEA, which are summarized in the following sections.
5.3.1. Filling Line Setup Activities:
A few considerations, best practices, improvements, and controls are listed below for filling line setup:
Transition to isolator technology to reduce the number of routes of particle introduction
Improve filling line design to simplify the line setup process
Use single-use disposable components and commodities where possible
Implement robust cleaning and decontamination processes
Put in place robust and detailed line setup procedures
Put in place robust operator training and qualification programs
5.3.2. Entry of Commodities and Materials into the Filling Space:
Implementation of robust control measures for entry of commodities and materials into the filling space can include some of the following activities:
robust decontamination procedures;
e-beam technology; and
short-cycle vaporized hydrogen peroxide (VHP) decontamination process.
A robust filling-line design can also minimize particle introduction. These activities would include controls around alpha-beta ports, direct integration of line with sterilizers, VHP chambers, and secondary processing equipment such as lyophilizers.
5.3.3. Interventions in the Filling Process:
A robust filling-line design, including isolator or RABS design, can minimize the need for operator intervention. Implementation of a robust ergonomic filling-line design that facilitates operator interventions where they are necessary and use of single-use disposable materials for interventions can also minimize introduction of particles. Robust process designs can minimize potential impacts of environmental monitoring, especially viable monitoring along with use of rapid online technologies.
5.3.4. Preparation, Storage, and Handling of Materials and Commodities:
The use of single-use disposable materials can help minimize the introduction of particle matter. Vigorous processes in preparation, storage, and handling of materials and commodities can include some of the following processes:
robust wrapping and particle control/elimination strategies;
robust transport and handling procedures; and
robust sterile holding-period validation processes.
5.3.5. Vendor Qualification Programs and Personnel:
Multiple factors can be improved, and vendor qualification/certification programs and personnel management/education are among those factors. A few considerations when taking into account vendors and personnel can include the following:
Robust quality technical agreements
Ensure robust incoming material and commodity testing/monitoring programs
Training programs for aseptic personnel
Ensure a robust requalification program for aseptic personnel
Gowning qualification program for aseptic personnel
6. Recommendations and Conclusion
It is critical to establish additional measurement sensitivity to better understand the sources and number of particles in the final product container after filling and sealing. Information sharing should also be a primary goal across workstreams developing analytical methods for particle detection, to enhance method sensitivity to the visible particle-size threshold. Additional considerations may be as follow:
Review and consider filling process design improvements and best practices to reduce particle contamination of final product containers.
Review and consider operator training and qualification best practices to reduce particle generation, transfer, and contamination during the filling operation.
Review and consider process improvements and best practices to reduce particle generation, transfer, and contamination in key process steps with consideration for protection of intellectual property of representative companies.
Engage RTU secondary component manufacturers to ensure universal best practices for packaging, handling, and use of trays, tubs, Tyvek sheets, and bags for packaging. Develop universal specifications and methods for evaluating cleanliness of such components.
Consider viable action items and/or changes to the process resulting from a review and consideration of process improvements.
Elaborate best practices to reduce particle generation and/or contamination in key process steps with consideration for protection of intellectual property of representative companies.
Establish additional measurement sensitivity to better understand the sources and number of particles in the container following critical steps. Ensure compatibility of methods to incoming materials at the glass and elastomer manufacturer.
Consider evaluation of incoming tube washing or additional glass-washing on the final particle-load.
The provided FMEAs can serve as a springboard that companies can use to start or continue their journey of continuous improvement and further reduce the probability of introducing particles throughout their manufacturing process. In addition, the FMEAs help to cultivate a deeper understanding of the supplier processes and to elaborate joint efforts to tackle the high need of industry to reduce the particle load.
A cross-functional team composed of glass suppliers, elastomer suppliers, and fill finish operations performed these FMEAs to identify areas across the end-to-end manufacturing process for visible particle entry routes into the final drug product. Thereby, the focus was placed on components widely used in industry as well as accounting for the best-in-class available processes and materials.
Processes posing the highest risk for deposition of visible particulate onto the surface of components or into container systems of injectable pharmaceutical product were thereby identified. These areas now provide a robust foundation from which to define best practices on how to reduce particles during manufacturing of parenteral drug formulations.
Common areas across the end-to-end manufacturing process were identified as particle entry routes, ranging from cleanroom protocols (line setup, operator intervention or cleanroom wipes), incoming packaging materials (i.e., TYVEK bags, nest, tubing, and so forth) as well as handling of materials throughout the process (i.e., glass-to-glass contact, transfer of material across clean rooms).
In addition, specific areas of manufacturing processes that allow for continuous improvement actions in future were detected. Glass cutting during glass manufacturing, cutting of panels or trimming of stoppers, as well as the right choice of manufacturing equipment are just some of the major areas that show potential to reduce particle load in final drug products in future.
Throughout upcoming activities within the workstream, data robustness needs to be established around the data provided in the FMEAs. The provided results will allow elaboration of best practices for some of the high-risk areas, which is an important piece throughout the ongoing activities in continuous improvement across the industry. As case studies are developed, the information will be combined into a comprehensive technical report.
Footnotes
PDA PAPER DISCLAIMER: The following paper is a special contribution from the Parenteral Drug Association (PDA). This article was internally reviewed by PDA and the task force members and not peer-reviewed by the PDA Journal of Pharmaceutical Science and Technology. This PDA paper is protected by copyright and unauthorized distribution or use is prohibited.
- © PDA, Inc. 2019
Additional Reading:
- 1.
- 2.
- 3.
- 4.
- 5.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}