
Abstract
Appropriate segregation within manufacturing facilities is required by regulators and utilized by manufacturers to ensure that the final product has not been contaminated with (a) adventitious viruses, (b) another pre-/postviral clearance fraction of the same product, or (c) another product processed in the same facility. However, there is no consensus on what constitutes appropriate facility segregation to minimize these risks. In part, this is due to the fact that a wide variety of manufacturing facilities and operational practices exist, including single-product and multiproduct manufacturing, using traditional segregation strategies with separate rooms for specific operations that may use stainless steel or disposable equipment to more modern ballroom-style operations that use mostly disposable equipment (i.e., pre- and postviral clearance manufacturing operations are not physically segregated by walls). Further, consensus is lacking around basic definitions and approaches related to facility segregation. For example, given that several unit operations provide assurance of virus clearance during downstream processing, how does one define pre- and postviral clearance and at which point(s) should a viral segregation barrier be introduced? What is a “functionally closed” system? How can interventions be conducted so that the system remains functionally closed? How can functionally closed systems be used to adequately isolate a product stream and ensure its safety? To address these issues, the member companies of the Consortium on Adventitious Agent Contamination in Biomanufacturing (CAACB) have conducted a facility segregation project with the following goals: define “pre- and postviral clearance zones” and “pre- and postviral clearance materials”; define “functionally closed” manufacturing systems; and identify an array of facility segregation approaches that are used for the safe and effective production of recombinant biologics as well as plasma products. This article reflects the current thinking from this collaborative endeavor.
LAY ABSTRACT: Operations in biopharmaceutical manufacturing are segregated to ensure that the final product has not been contaminated with adventitious viruses, another fraction of the same product, or with another product from within the same facility. Yet there is no consensus understanding of what appropriate facility segregation looks like. There are a wide variety of manufacturing facilities and operational practices. There are existing facilities with separate rooms and more modern approaches that use disposable equipment in an open ballroom without walls. There is also no agreement on basic definitions and approaches related to facility segregation approaches. For example, many would like to claim that their manufacturing process is functionally closed, yet exactly how a functionally closed system may be defined is not clear. To address this, the member companies of the Consortium on Adventitious Agent Contamination in Biomanufacturing (CAACB) have conducted a project with the goal of defining important manufacturing terms relevant to designing an appropriately segregated facility and identifying different facility segregation approaches that are used for the safe and effective production of recombinant biologics as well as plasma products.
- Functionally closed
- Viral clearance
- Facility segregation
- Adventitious agent contamination
- Physical segregation
- Open ballroom
Introduction
It is expected that recombinant biopharmaceutical and plasma products will be manufactured in a way that ensures their safety with regard to absence of viral contaminations. This is generally accomplished using a three-tiered approach: selection of starting and raw materials that have a low risk of containing adventitious virus; testing of cell banks and in-process materials to ensure they are virus-free; and incorporation of steps during product purification to remove or inactivate potential adventitious or endogenous viral contaminants (1). We would note that emerging technologies and regulatory requirements may mean additional steps need to be taken to assure viral safety. Another key aspect of a biopharmaceutical manufacturing facility's overall viral safety strategy is the use of segregation within the facility to ensure that product streams are not inadvertently contaminated with viruses or other products in the manufacturing facility. This is a regulatory expectation; however, because effective facility segregation may be context-dependent, specific expectations in regulatory guidance documents are lacking. For example, the World Health Organization (WHO) GMP for Biological Products (2) contains the following: “In cases where a viral inactivation or removal process is performed, measures (e.g., related to facility layout, unidirectional flow, and equipment) should be taken to avoid the risk of recontamination of treated products by non-treated products.” Similarly, Section XVIII.E of the United States GMP Guidance for Active Pharmaceutical Ingredients (3) states: “Appropriate precautions should be taken to prevent potential viral contamination from previral to postviral removal/inactivation steps. Therefore, open processing should be performed in areas that are separate from other processing activities and have separate air handling units.” Other guidance documents touch on facility segregation as well (4⇓⇓⇓–8).
This lack of clarity in documented regulatory expectations means that standards are often, at least in part, driven by industry itself. One company may receive a regulatory observation after implementing a specific facility segregation approach. Given the time and economic pressures to see a product attain commercial viability, rather than engage with the authorities regarding the acceptability of the specific approach, companies will often choose to implement a more conservative approach to facility segregation. Thus, one company's implementation of a conservative approach sets a bar that subsequent companies are expected to meet.
At the same time, there is a strong desire by industry to develop new, more cost-effective facilities. While there is proven acceptance of existing facility design (e.g., physically segregating pre- and postviral processes in separate rooms), such approaches can be expensive. In contrast, using disposable equipment and open ballroom designs, where multiple processing steps occur in the same room without being physically segregated, can reduce manufacturing facility costs but has a higher perceived risk due to lack of experience.
Further, it is practically impossible for one company to institute a global, harmonized approach to facility segregation. First, differences between manufacturing facilities, both existing and new, may preclude the use of a uniform segregation approach. Second, segregation approaches used in new processes may not be possible to implement in legacy processes. Third, different products may have specific processing requirements and different intrinsic degrees of contamination risk (e.g., plasma derivatives versus recombinant products manufactured from cell culture) that necessitate the use of different facility segregation approaches.
Lastly, consensus is lacking around basic definitions related to facility segregation, including functionally closed systems and pre- and postviral clearance. One proposed segregation approach is the idea of a “functionally closed” system that effectively isolates the product stream while allowing open manipulations. While it would be beneficial to argue that a functionally closed system would provide an adequate level of segregation, there is disagreement about how such a system is defined. For example, Palberg et al. define a functionally closed system as “a process system that may be routinely opened (e.g., to install a filter or make a connection) but is returned to a closed state through a sanitization or sterilization step prior to process use,” (9) whereas Odum et al. define it as “a system that when operated under normal conditions prevents the ingress or egress of adventitious agents” (10). The International Society for Pharmaceutical Engineering (ISPE) Baseline Guide Volume 6 also defines functionally closed using language such as “… appropriate measures have been exercised to render the system closed” (11). Further, the ISPE definition requires that processes are validated as functionally closed, whereas Phase I/II clinical manufacturing may not be validated. Therefore, there is a need for a definition of functionally closed that is flexible enough to apply to all manufacturing stages and arrived at by consensus. Consensus is also lacking around how one defines pre- and postviral clearance. Viral clearance is a continuum throughout the downstream process, and only by combining multiple steps can an overall viral clearance claim be reached. Given this, how does one decide where to implement segregation of pre- and postviral clearance process streams?
Both the industry and the regulatory authorities desire the predictable and certain supply of biopharmaceuticals to meet patient need. A consistent and clearly defined approach to the principles and objectives of facility segregation would support this goal; yet, the challenges listed above make such a clearly defined approach difficult to achieve. To address this issue, the Consortium on Adventitious Agent Contamination in Biomanufacturing (CAACB), a biopharmaceutical industry consortium housed at the Massachusetts Institute of Technology's (MIT) Center for Biomedical Innovation, convened experts from its member companies with the following goals:
Reach a consensus around definitions of common facility segregation terminology, specifically:
pre- and postviral clearance zones;
pre- and postviral clearance process materials; and
functionally closed manufacturing systems
Agree-upon appropriate facility segregation approaches.
The following presents the status of the work to date.
Approach
The CAACB was formed in January 2011 as an initiative at MIT's Center for Biomedical Innovation. As of publication, the CAACB consists of 24 member companies. The broad mission of the CAACB is to pool biomanufacturing expertise and data in the area of adventitious agent contamination. The consortium provides a forum for discussion of adventitious agent contamination experiences, risk of contamination, and methods to mitigate risks. In the context of this effort, the CAACB convened a working group consisting of experts from each member company that manufactures biopharmaceuticals. The working group first discussed and agreed upon definitions of the following terms: previral clearance zone, postviral clearance zone, previral clearance material, postviral clearance material, and functionally closed manufacturing system. The working group next identified and agreed upon an array of facility segregation approaches that are suitable and appropriate for the manufacture of safe and effective biologics.
The scope of this work is intended to address facility segregation for viral safety in the production of recombinant biologics as well as plasma products, which includes the following: approaches to prevent cross-contamination between products; approaches to prevent contamination of postviral clearance material with previral clearance material; and approaches to prevent contamination to, or from, the environment or personnel. The concepts discussed in this paper should be applicable to a wide array of manufacturing situations, including for both multiproduct and single-product facilities, as well as facilities that use stainless steel equipment and those that use single-use equipment. There will be some overlap in the concepts presented below regarding the controls used for segregation for viral safety and those used for microbial safety.
Definition of Pre- and Postviral Clearance
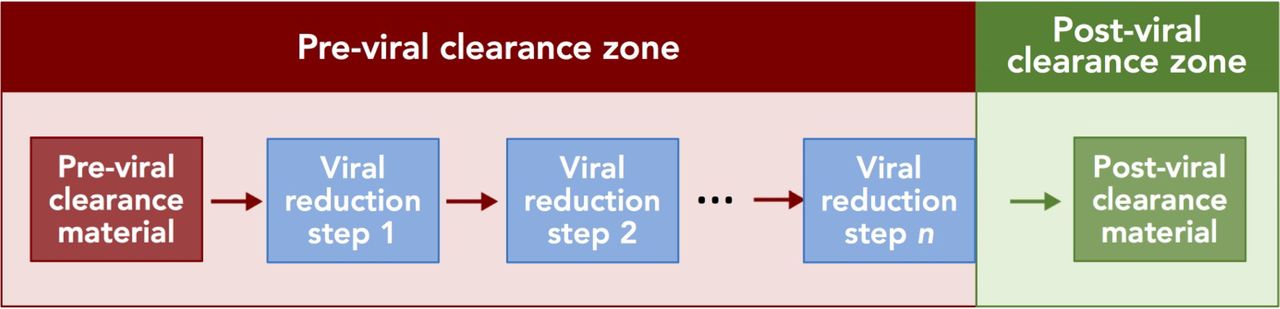
As noted above, viral clearance is a continuum throughout the downstream process, and the total claimed clearance capacity of a process relies on several unit operations together. With this in mind, the following definitions have been developed (Figure 1): material that is claimed to be postviral clearance material is material (e.g., a product stream) that has been processed through one or more demonstrated virus reduction unit operations; material that is claimed to be previral clearance material is material (e.g., a product stream) that has not been processed through the same virus reduction unit operations; areas of the facility or process claimed to be postviral clearance zones are part of the manufacturing process downstream of the planned, demonstrated virus reduction unit operations; and areas of the facility or process claimed to be previral clearance zones are the part of the manufacturing process upstream of the same virus reduction unit operations. Finally, a demonstrated virus reduction unit operation is one in which phase-appropriate data are used to support the viral clearance capability of a specific unit operation and may be dedicated for virus reduction. In the case of early-stage clinical manufacture, viral clearance data from a different, but similar, product may be sufficient to demonstrate the clearance capacity, whereas for commercial manufacture, the viral clearance capacity of the respective unit operations will be validated. Common examples of demonstrated virus reduction unit operations include, but are not limited to, nanofiltration (12, 13), solvent/detergent (S/D) treatment (14, 15), chromatography (16), and low pH inactivation (17, 18).

Schematic representation of pre- and postviral clearance zones and material. Material that is claimed to be previral clearance material is processed through one or more demonstrated virus reduction unit operations (blue boxes, labeled step 1 through step n). Red arrows indicate previral clearance material; the green arrow indicates postviral clearance material; the red box indicates the previral clearance zone; and the green box indicates the postviral clearance zone.
Postviral clearance material should be handled such that the risk of cross-contamination from previral clearance material has been reduced to a level determined to be acceptable by the manufacturer. To reduce the risk of cross-contamination of postviral clearance material with previral clearance material, the previral clearance zone should be segregated from the postviral clearance zone through approaches that may include a combination of physical segregation, segregation by time (temporal segregation), procedural segregation, the use of functionally closed systems, cleaning and disinfection, and appropriate engineering controls. Segregation of pre- and postviral clearance material may be applied once (e.g., after the last viral clearance unit operation) or to multiple viral clearance unit operations as necessary depending upon the facility, equipment, product type, and production process. It should be noted that in the case of segregation with physically separate rooms, it is often impossible for the virus reduction step to occur exactly at the room boundary, and postviral clearance material will spend some time in the same room as previral clearance material; therefore, the controls in place to maintain segregation between the previral clearance material and the postviral clearance material in the same zone should take this into account.
Industry Approaches to Facility Segregation
With the above definition of pre- and postviral clearance, it is now possible to identify an array of facility segregation approaches that may be used for the manufacture of safe and effective biopharmaceuticals. We note that the approaches to facility segregation outlined below are based on the following assumptions:
Replication of an adventitious viral contaminant or production of endogenous viral particles during manufacturing operations can occur in only upstream cell culture operations. Therefore, the only routes of potential virus ingress that are considered of sufficient risk to the process are through a tissue or cell culture starting material (e.g., plasma, cell lines), a cell culture raw material (e.g., media or media components), or manufacturing personnel in cell culture.
Personnel and raw materials (e.g., buffers and chromatography resins) used in downstream operations are not considered a significant potential source of virus ingress, as downstream operations provide no opportunity for virus proliferation. However, it is acknowledged that the risk of introducing a virus into downstream operations is not zero.
The risk of contamination from each raw material, including any used in downstream operations, has been assessed, and for raw materials that are deemed to be of sufficiently high risk, appropriate measures have been taken to reduce that risk (e.g., the use of effective virus reduction methods to reduce the virus contamination risk from the high-risk material, replacing the high-risk material with one of lower risk, or testing the raw material for viruses).
When cleaning or sterilization is used to either close a system or segregate manufacturing processes that use the same nondedicated, reusable equipment, the effectiveness of the cleaning or sanitizing agents has been demonstrated through a combination of the following options:
studies of the viricidal effectiveness of the cleaning or disinfection procedure;
literature demonstrating the effectiveness of the cleaning agent or disinfectant;
data from the supplier of the cleaner or disinfectant supporting its efficacy; and
steam-in-place (SIP) validation based on temperature-resistant challenge species, such as bacterial spores.
Comprehensive pest control practices are used.
Viral reduction steps are demonstrated to effectively remove or inactivate virus.
Processes are operated under GMP.
The term physical segregation refers to separation of previral clearance and postviral clearance zones by walls with either separate heating, ventilation, and air conditioning (HVAC) or single-pass air and pressure difference, including dedicated access for personnel with dedicated gowning, where gowning rooms function as airlocks.
Controls are used during operations, specifically after the last demonstrated virus reduction unit operation, to ensure that the risk of cross-contamination or introduction of a virus has been reduced to a level determined to be acceptable by the manufacturer.
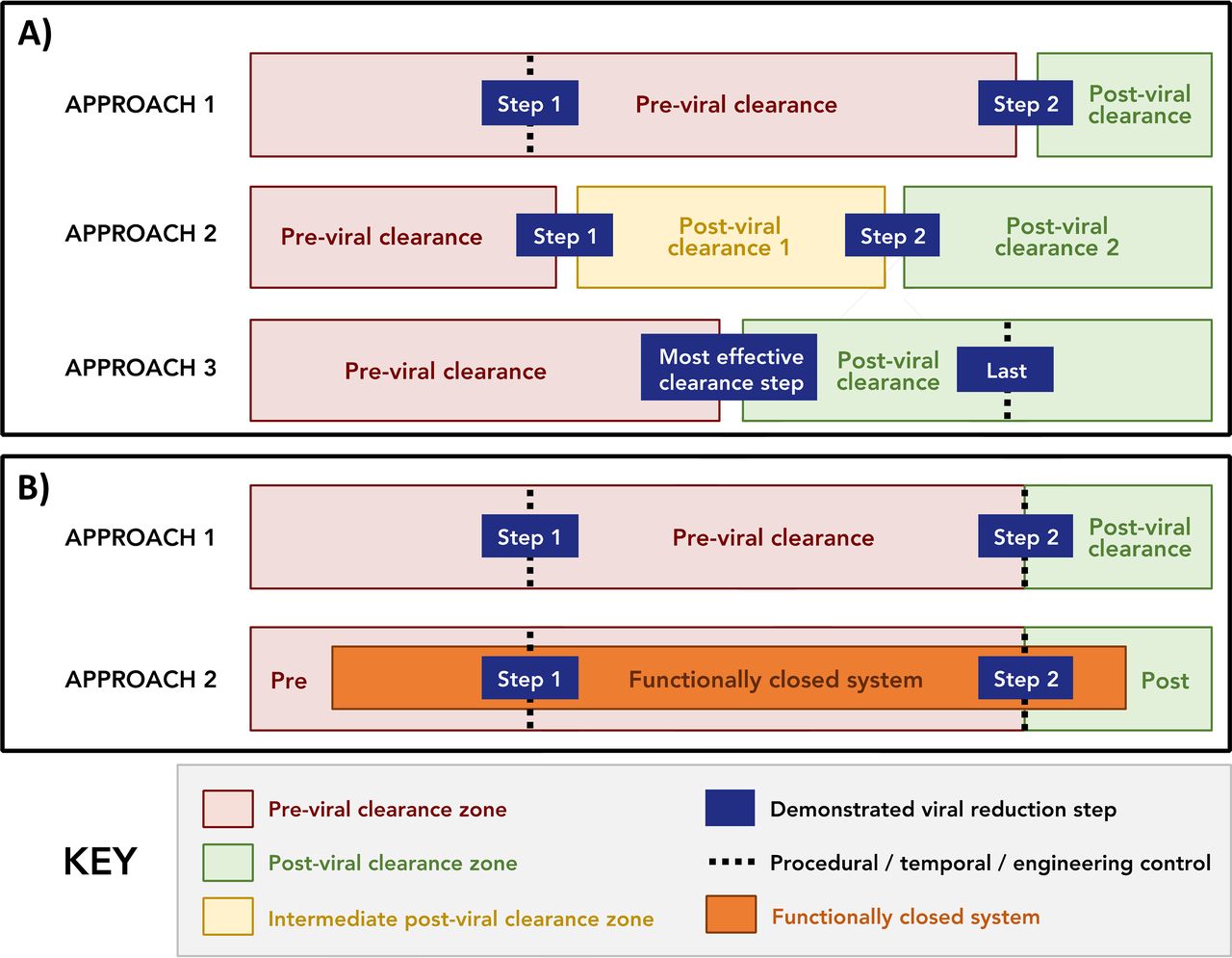
Based upon the definitions and assumptions listed above, two broad classes of facility segregation were identified as approaches that use (a) physical segregation and (b) open ballrooms, which we define as a room/area with multiple unit operations that uses segregation approaches other than physical segregation. These are schematically represented in Figure 2. It should be noted that Figure 2 attempts to capture only the major classes of facility segregation and that hybrid approaches are also acceptable, provided that appropriate controls are employed. It should also be noted that the options listed below are applicable to both single-use and multi-use facilities.

Acceptable approaches to facility segregation.
A) Facility segregation approaches that utilize physical segregation have been broken down into three distinct variations: approach 1, where physical segregation is used around the last demonstrated viral clearance step; approach 2, where physical segregation is used around multiple (at least two) demonstrated viral clearance steps; and approach 3, where physical segregation is used around the “most effective viral clearance step” (Note: when the last step is also the most effective, approach 3 is functionally identical to approach 1). B) Facility segregation approaches that utilize ballroom operations can be split into two distinct approaches: approach 1, where ballroom-style operations use traditional equipment, manufacturing operations occur in the same room, and physical segregation approaches, as previously defined, are not used; and approach 2 with ballroom operations utilizing functionally closed systems. Note: in both A) and B), gaps between bars indicate that these areas are physically segregated, as defined earlier in this document, and dashed lines indicate that process streams upstream of the effective viral clearance step are segregated by temporal, procedural, and/or engineering controls.
Physical Segregation Approaches and Their Controls
Manufacturing approaches that use physical segregation are well understood and accepted within the industry. As such, three distinct, but related, variations of facility segregation were identified where the previral clearance zone and the postviral clearance zone are physically segregated (Figure 2A). Specifically, in physical segregation approach 1, segregation is used around the last demonstrated viral clearance unit operation; in approach 2, physical segregation is used around multiple (at least two) demonstrated viral clearance unit operations; and in approach 3, physical segregation is used around the “most effective viral clearance unit operation,” where the term “most effective” is in relation to all other viral clearance unit operations in the process. In addition, we would note that there are guidance documents that define the minimum acceptable viral clearance that may be claimed as effective (19). In the case of physical segregation approach 3, the most effective viral clearance unit operation should be determined by the manufacturer using a predefined procedure, such as validated data or a risk assessment.
For all three physical segregation approaches in Figure 2A, it was determined that the following controls are necessary to ensure that the previral clearance zone and postviral clearance zone remain segregated:
There are dedicated personnel and material airlocks for pre- and postviral clearance zones. Depending on the exact facility design, these may be separate or combined airlocks.
Product-contacting equipment is dedicated for either a specific product or a single use, or an effective sanitization procedure is used to clean and sanitize the equipment prior to re-use.
Personnel, materials, equipment, and waste flows are procedurally controlled to avoid the contamination of postviral clearance materials with previral clearance materials.
Dedicated water for pre- and postviral clearance zones is not necessary, provided controls are in place to prevent contamination.
Dedicated wash areas for pre- and postviral clearance zones are not necessary, provided the risk of cross-contamination through the wash area is effectively mitigated through appropriate controls, such as sanitization, temporal segregation, etc.
For drainage and wastewater, there are controls to prevent backflow.
Temporal, procedural, and/or engineering controls are used to prevent external/cross-contamination of intermediates before and after other viral reduction steps.
It should be noted that approach 3 is equivalent to approach 1 when the most effective virus reduction unit operation is also the last virus reduction unit operation. However, when this is not the case, as depicted in Figure 2A, additional controls need to be used around the last viral clearance unit operation, as there are no additional viral clearance steps in the process to ensure product safety. These decisions should be driven by a risk assessment.
Ballroom Approaches and Their Controls
As noted previously, there is significant interest in the biopharmaceutical industry in approaches that use open ballrooms, defined earlier, as they may allow decreased costs and reduce the time for a facility to start up and get running (10, 20). Figure 2B identifies two facility segregation approaches that use open ballrooms: ballroom approach 1, where pre- and postviral clearance zones are in the same room and are segregated through temporal, procedural, and/or engineering controls to prevent external or cross-contamination of postviral clearance material, and ballroom approach 2, which leverages functionally closed manufacturing systems to ensure that pre- and postviral zones are segregated.
In the case of ballroom approach 1, the following controls should be used to ensure that the previral clearance zone and the postviral clearance zone remain segregated:
Equipment that comes into contact with product is dedicated for either a specific product or is single use, or an effective sanitization procedure is used to clean and sanitize the equipment prior to reuse.
Dedicated wash areas for pre- and postviral clearance zones are not necessary, provided the risk of cross-contamination through the wash area is effectively mitigated through appropriate controls, such as sanitization, segregation by time, etc.
For drainage and wastewater, there are controls to prevent backflow.
Personnel, materials, equipment, product flows, and waste flows within the facility are procedurally or temporally controlled. One example of a procedural control is unidirectional flow.
Risks from open operations, such as open-column packing, are controlled via procedural or temporal controls.
In the case of ballroom approach 1, segregation is largely attained via procedural, temporal, or engineering controls. Below are some specific examples of these controls.
Example 1: In a room where there is open handling of previral clearance material and where postviral clearance material is also processed, such as filter integrity testing of a filter used upstream of a S/D treatment step, the following controls may be used:
During S/D treatment in a closed tank, the in-line filter between the mixing tank and the inactivation tank is flushed with water for injection (WFI) and tested for filter integrity before it is removed. Flushing with WFI before filter removal minimizes the risk of pretreatment material (potential source of viral contamination) escaping into the environment.
Single-use coats and gloves are worn by the operators during filter removal and are discarded immediately afterward.
Room access during filter removal is restricted to only the operators involved.
A sick employee policy is implemented to prevent virus ingress from personnel.
Example 2: When packing a chromatography column used for postviral clearance material in a previral clearance zone, the following controls may be used:
If the chromatography resin is compatible with harsh cleaning and regeneration (e.g., with NaOH at an appropriate concentration and contact time) that is also proven to be effective for virus inactivation, no additional controls are necessary, as the regeneration of the chromatography resin prior to use will serve as an effective segregation.
If the chromatography resin is not compatible with cleaning and regeneration that has been proven to be effective for virus inactivation, then the resin and packing operation must be protected from sources of potential viral load. The column packing, or unpacking, may be performed in a separate room that is kept closed throughout the manipulation with no other concurrent open operations in the same room. Column and packing hardware must be cleaned with proven viral effective chemicals or be of sterile and single-use type. Column packing status is prominently labeled and access to the room is also restricted while column packing operations are underway. Single-use coats, facemasks, and gloves are worn by the operators conducting column packing or unpacking operations. A sick employee policy is implemented to prevent virus ingress from personnel.
Example 3: A normal flow virus filter is validated for retrovirus removal in the manufacturing process but cannot be steamed or base sanitized. In addition, the filter housing must be removed to introduce the filters prior to flushing, integrity testing, and product load. The following controls may be used:
Prior to the introduction of product feed stream, the entire flow path can be sanitized while bypassing the viral filter with a solution having demonstrated viricidal properties under the conditions of use.
Closed operations and gowning procedures (e.g., glove changes prior to handling the virus filter), together with spatial and personnel controls, to control for cross-contamination risk from other open operations (e.g., Protein A column unpacking) may be used during processing of the product stream.
In the case of ballroom approach 2, pre- and postviral clearance material is processed in the same room, and the use of functionally closed manufacturing systems adequately prevents external or cross-contamination of the postviral clearance material. Additional controls that should also be used include:
Product flow through the functionally closed manufacturing process is unidirectional.
Product-contacting equipment is dedicated for either a specific product or single use, or an effective sanitization procedure is used to clean and sanitize the equipment prior to reuse.
Procedural and engineering controls are used to ensure that the manufacturing process remains functionally closed during addition or removal of material or equipment from the process.
In the case of ballroom approach 2, adequate facility segregation hinges almost entirely on the use of functionally closed manufacturing. Therefore, it is important to have a robust, consensus definition of what constitutes functionally closed as well as understand the potential implications of employing such systems. Some key questions that must be addressed when considering a functionally closed system include the following: Can open operations be performed on a functionally closed system? Can material transfers into or out of a functionally closed system be performed? How do you demonstrate that a manufacturing system is functionally closed? How do you demonstrate that a manufacturing system is functionally closed during operation? What are the consequences of a breach in a functionally closed system? The following section presents the work of the CAACB with regard to these issues.
Definition of a Functionally Closed System
The CAACB Facility Segregation Working Group has reached consensus on the following definition of a functionally closed system: A functionally closed system is one that, when in operation, isolates the process stream from the manufacturing environment. A functionally closed system must be demonstrated to show that the risk of contamination (e.g., bacterial, viral, and other products in the same facility) from or to the manufacturing environment has been reduced to a level determined to be acceptable by the manufacturer such that the room, manufacturing personnel, and other concurrent manufacturing operations are a noncritical aspect of the product.
It is understood that there is always some level of risk when performing biopharmaceutical manufacturing operations. Therefore, the goal should be to reduce residual risk to an acceptable level, and the definition of functionally closed was developed with that in mind.
Having developed the definition of a functionally closed system, the questions posed above can now be considered in more detail. First, can open operations be performed on a system that is claimed to be functionally closed? Ideally, a functionally closed system would be designed such that it need not be opened during routine processing. However, should planned open operations be deemed necessary, it must also be demonstrated to meet the definition of functionally closed. As an example, an open operation should be designed such that the process stream remains isolated from the manufacturing environment while the system, or a portion of the system, is opened for the planned operation. The portion of the system that was exposed to the manufacturing environment may then be returned to a closed state through an appropriate and validated sterilization or sanitization procedure.
Second, can material transfers into or out of a functionally closed system be performed? As with open operations, transfers into or out of a functionally closed system may be performed only if they also have been demonstrated as functionally closed (e.g., the risk of contamination from or to the manufacturing environment has been reduced to a level determined to be acceptable by the manufacturer such that the room, manufacturing personnel, and other concurrent manufacturing operations are a noncritical aspect of the product); otherwise, such a transfer would mean that the system is no longer considered to be functionally closed.
The CAACB has identified two specific examples of material transfers that meet the definition of functionally closed. First, a material transfer may be performed through connections that are functionally closed. Some common examples include the use of aseptic connectors or sterilization or sanitization after connection to reduce potential contaminants below a level determined to be acceptable by the manufacturer. Second, the definition of functionally closed is related to reducing the risk of contamination to an acceptable level. Therefore, the following meets the above definition as well: the transfer of raw materials deemed to be of low risk for virus contamination (e.g., a risk assessment has determined that the raw material is a low risk of contamination or an inactivation or removal step has been applied to a high-risk material) into, or out of, a system through a functionally closed sterilizing-grade filter manifold.
Given the above definition of functionally closed and the related considerations for open operations and material transfers, the following are real-world examples of procedural, temporal, or engineering controls that can be used when in ballroom approach 2 to ensure appropriate segregation.
Example 1: The product stream enters a single-use, gamma-irradiated processing bag via an aseptic connection. The low pH viral inactivation solution is added through an aseptic connection until the inactivation pH range is achieved. The adjusted pool may be transferred to another bag via aseptic connection where the inactivation hold time occurs. The decision to perform this transfer is informed by an appropriate risk assessment. When the viral inactivation hold time is achieved, the product is transferred into the next functionally closed operation through an aseptic connection. The pre-inactivation equipment and single-use components remain functionally closed, i.e., are disconnected aseptically and remain isolated from the manufacturing environment, and are removed from the area for disposal.
Example 2: Final bagging of the postviral clearance drug substance occurs in the same open room as all preceding previral clearance activities. The filling manifold is assembled using gamma-irradiated components and aseptic connections. The sterilizing-grade filter is gamma irradiated with aseptic connectors on the inlet and outlet so the filter and manifold can be connected while maintaining a functionally closed system. Each bag to be filled is connected via tube welding or an aseptic connector, which enables the connection of two separate fluid pathways while preventing the introduction of environmental contaminants into those pathways.
Example 3: When viral filtration operation occurs in an open room where previral clearance operations have occurred or are ongoing, the following controls are used:
If the virus filter is compatible with treatment using a harsh cleaning agent, such as NaOH, the entire flow path, including the filter, may be sanitized using proven effective conditions with NaOH after assembly to close the system. A sterile bag is connected to the filter outlet via aseptic connectors to create a functionally closed system through the viral filtration operation.
If the virus filter is not compatible with treatment using a harsh cleaning agent, then the filter must be protected from sources of potential viral load. The filter assembly may be performed in a separate room that is kept closed throughout assembly and integrity testing. No other concurrent open operations occur in the same room. Hardware must be cleaned with proven viral effective chemicals or be of sterile and single-use type. Filter assembly status is prominently labeled. While filter assembly operations are underway, access to the room is also restricted. The filtrate side of the filter must be protected to ensure it remains functionally closed during installation in the previral clearance zone (e.g., via use of aseptic connections). Single-use coats, facemasks, and gloves should be worn by the operators conducting filter assembly operations.
Example 4: All buffers that flow into the chromatography system pass through sterilizing-grade filters to protect against microbial contamination. These filters are not designed to remove virus particles. However, with the appropriate procedural controls, these solutions are viewed as a very low risk for virus ingress, as in the absence of mammalian cells, there is no opportunity for virus replication and also provided that (1) they are not of animal or human origin (and have been appropriately segregated from animal origin material all along the material supply chain, including during material weighing and dispensing steps) and (2) high-risk materials have been treated to eliminate any significant risk.
Demonstrating that a System is Functionally Closed
When designing a functionally closed system, it is necessary to demonstrate that the system, as designed, is actually closed. There are crucial questions about how to approach this issue. The CAACB Facility Segregation Working Group has identified the following examples of how a system may be demonstrated to be closed. It is noted that multiple methods may be used to demonstrate that a system is closed and that the standard for demonstrating closure should be commensurate with the associated risk of viral load. For example, as cell culture can amplify virus, operations that involve the growth of cells all the way through to harvested cell culture fluid pool hold have the highest risk and will require the highest demonstration of closure.
Validating that the sanitization or sterilization procedure that is used to return the equipment to a closed state removes potential carryover from the previous process stream as well as other relevant biological contaminants (e.g., kill of biological indicators or inactivation data of representative target or model viruses) below levels determined to be acceptable by the manufacturer.
Demonstrating that the defined process boundary of the system contains the process stream and provides a barrier to ingress of environmental contaminants. Some examples of such a demonstration include:
Integrity testing to demonstrate the integrity of the defined process boundary, for example, validated integrity testing of filtration operations, a pressure hold test, a validated SIP performed under pressure, and/or a helium leak test. As the integrity of the process barrier is imperative to maintaining functional closure, integrity testing should be performed if the equipment is stressed during the process step such that the integrity of the equipment comes into question.
A more extreme example would be a media challenge test where the system is initially returned to a closed state (the process boundary is closed and the system is sanitized or sterilized using a validated procedure), charged with media using a closed transfer, incubated under relevant processing conditions (e.g., process relevant temperature and time), and then tested for biological contamination.
In addition to demonstrating that the system as designed is closed, it is also necessary to ensure that the system is returned to a closed state after installation and remains closed while in operation. The following are examples of how a system may be demonstrated, operationally, to be closed; multiple methods may be used to demonstrate that a system is closed:
The proper equipment is installed and used following a predefined sterilization or sanitization procedure, and no unwanted events or phenomena were observed during its installation or operation.
Verifying that the validated sanitization and sterilization procedures that were used to return the system to a closed state reached the targeted sanitizer concentration, contact times, and temperatures through proper observation and monitoring.
In the case of single-use equipment, verifying that the equipment manufacturer sterilized the system using a validated procedure (e.g., gamma irradiation) and that the system was installed using approved operating procedures that maintain the system as closed.
After the system is installed or returned to a closed state, performing an appropriate pre-use integrity test (e.g., pressure hold or helium-leak testing).
Testing of samples with an appropriately sensitive and specific test method from the process stream to verify that no contaminants (e.g., bacterial, viral, or other products) were detected during or after the closed manufacturing operation.
During processing, some types of equipment (e.g., chromatography skids) can be maintained under positive pressure, and the pressure is monitored (e.g., alarm-controlled) as a control of the integrity of the system.
Observation and visual inspection of the system to identify and address leaks.
Consequences of a Breach in a Functionally Closed System
Finally, it is important to consider the implications of a breach of a functionally closed system in a number of potential manufacturing scenarios. Thinking through these scenarios is necessary to understand the impact of a breach and the level of room classification that may be necessary to ensure an adequate safety margin. Four different scenarios were identified and considered in detail and are presented below. However, for all four scenarios, there are three specific risks to consider due to the breach for each scenario: the risk to the process stream, the risk to operators, and the risk to other manufacturing operations. In the case of the risk to the process stream, the exact risk will be scenario dependent. However, in all four scenarios, if the process stream is not contained, then the spill would need to be cleaned and disinfected using a validated method to reduce the risk of cross-contamination, and the process system would need to be sanitized (or replaced in the case of presterilized components) to be brought back into a closed state. In addition, in all four scenarios, the risk to operators will be dependent on the contents of the process stream and the stage of manufacturing (as drug potency often increases with purification). Regardless, a procedure to ensure a timely spill response, including spill containment with restriction of flows and regowning owing to soiled garments, should be in place. Additional details on the four scenarios are presented in the following section.
The first scenario is a breach in a controlled and classified environment (e.g., ISO 7 or 8) with no other concurrent manufacturing or open operations or with other closed manufacturing operations in the same room. In this case, if the process stream is still contained in the manufacturing vessel, the risk of contamination of that process stream from the environment may be considered acceptable based on the room classification and appropriate in-process controls, e.g., additional monitoring of samples or particle measurements. However, this procedure should be based on a risk assessment. As part of the risk assessment, it should be evaluated if the segregation of pre- and postviral clearance material was maintained despite the breach. As there are no other open operations in this scenario, the risk to other process streams is minimal, provided the spill is adequately cleaned.
The second scenario is a breach in a controlled and classified environment (e.g., ISO 7 or 8) with other concurrent open operations. In this case, there is a risk of cross-contamination to the process stream from the other open manufacturing operations (including maintenance or system setup) and vice-versa, and the process stream may need to be discarded or reprocessed depending on an appropriate risk assessment. In the worst-case scenario, the risk is considered unacceptable, and one or both of the process streams would need to be discarded. Again, this procedure should be based on a risk assessment.
The third scenario is a breach in a controlled but not classified environment with no other concurrent manufacturing or with other closed manufacturing operations in the same room. In this case, if the process stream is still contained in the manufacturing vessel, the risk of contamination from the environment may be considered unacceptable, and the process stream would be discarded or reprocessed depending on an appropriate risk assessment. As there are no other open operations in this scenario, the risk to other process streams is minimal, provided that the spill is adequately cleaned and disinfected using a validated method to reduce the risk of cross-contamination.
Finally, the fourth scenario is a breach in a controlled but not classified environment with a concurrent manufacturing system that has also breached. This is the worst-case scenario, as it is unlikely that two manufacturing systems would be breached at the same time. However, in this case, owing to other open operations occurring at the same time, there is a risk of cross-contamination from the other breached manufacturing operation and the environment, and the process stream may need to be discarded or reprocessed depending on an appropriate risk assessment. In the worst-case scenario, the risk is considered unacceptable, and one or both of the process streams would need to be discarded.
The definition of functionally closed, as well as the above thought experiment, demonstrates that if a functionally closed system can be defined, validated, and operationally maintained, then operation should be possible in a manufacturing environment with reduced room classification. The two scenarios in controlled but not classified environments did not identify significant risks to other functionally closed manufacturing operations in the case of a breach.
Conclusions
It is clear that there is no one single approach to appropriate facility segregation. Approaches that use physical segregation are well understood and accepted and have been used effectively for many years to produce safe and effective biopharmaceutical products. Current biopharmaceutical industry trends are toward smaller, more flexible manufacturing facilities. Being able to leverage a robust, consensus definition of functionally closed systems will undoubtedly facilitate this transition. The use of a functionally closed system has a number of potential advantages. If a functionally closed system can be defined, validated, and maintained closed, then manufacturing operation should be possible in an environment with reduced room classification. This would reduce the burden on facility management and design, thereby reducing costs. In addition, the use of functionally closed systems is expected to increase manufacturing flexibility and facility utilization owing to the ability to manufacture multiple products in the same facility at the same time.
Despite these advantages, the use of a functionally closed system faces a number of challenges. First and foremost, there is no industry- and regulatory agency-wide acceptance of the concept of functionally closed. This is clear in the lack of consensus around a definition of what constitutes functionally closed. Second, the use of new technologies required for functionally closed manufacturing operations carries a higher level of risk than traditional approaches. The complete risks of such new technologies may not yet be fully understood or accepted by industry and regulators. There are also larger risks in assuring continuity of the supply chain for many of these disposable technologies, as there may be little control over polymeric raw materials used to manufacture disposable bags. Third, it is important to note that in the case of using a functionally closed manufacturing system in a controlled nonclassified space, there is no routine environmental monitoring. This would lead to higher risk from the manufacturing environment, as the existing flora will not be known, and it will be more challenging to prove that sanitization and cleaning procedures are effective against possible contaminants. Finally, as currently the absence of virus cannot be proven, this paper has presented a number of thoughts on how to demonstrate that a system is functionally closed. This paper should help address some of these challenges by establishing a consensus definition of functionally closed, as well as pre- and postviral clearance, from the biopharmaceutical manufacturers that are members of the CAACB.
Conflict of Interest Statement
Paul W. Barone, Flora J. Keumurian, James C. Leung, Michael E. Wiebe, and Stacy L. Springs declare they have no competing financial interests. All other authors declare that they are employees of their respective companies.
Acknowledgements
The authors would like to acknowledge the support of the members of the Consortium on Adventitious Agent Contamination in Biomanufacturing (CAACB). The current members of the CAACB include Amgen, Asahi Kasei Bioprocess, Biogen, BioMarin, Boehringer Ingelheim, Brammer Bio, Bristol-Myers Squibb, Charles River Labs, CSL Behring, Eli Lilly and Co., EMD Serono, Genentech, Histogenics, LFB, MedImmune, Merck & Co., MilliporeSigma, Pfizer, Regeneron, Sanofi, Sanofi Genzyme, Sanofi Pasteur, Shire, and ThermoFisher. The authors would like to thank the members of the CAACB Steering Committee for their review of, and feedback on, this manuscript.
- © PDA, Inc. 2019




{kind=link}
{kind=link}