
Abstract
The National Institute for Innovation in Manufacturing Biopharmaceuticals (NIIMBL) piloted a forum to encourage an exchange of information between the biopharmaceutical industry and the U.S. Food and Drug Administration (FDA). To facilitate this exchange, NIIMBL conducted a survey of industry representatives around the perceived challenges associated with the adoption of new innovative technologies for biopharmaceutical manufacturing or for continuous improvement and then held an Active Listening session with industry and FDA stakeholders to share common themes. The scope was limited to biotechnology products regulated by the Center for Drug Evaluation and Research (CDER). This manner of exchange has not been tested before and led to meaningful dialog between industry and the Agency and valuable takeaways by all involved. One of the general findings and key points of discussion was around the perceived lack of a business case for adoption of new technology in the manufacture of monoclonal antibodies and therapeutic proteins. Tight timelines were the primary constraints for hesitation around pre-approval implementation and the challenges associated with a global regulatory environment were the primary constraint around post-approval adoption of new technology. Mechanisms that would allow industry and regulatory scientists to develop a shared understanding of new technologies, outside of formal applications, could de-risk adoption of new technologies by the industry. The favorable response to this NIIMBL-facilitated exchange suggests that this format could be useful in establishing a more informal dialog between the FDA and industry on industry-wide challenges.
1. Introduction
The mission of the National Institute for Innovation in Manufacturing Biopharmaceuticals (NIIMBL) is to accelerate biopharmaceutical manufacturing innovation, support the development of standards that enable more efficient and rapid manufacturing capabilities, and educate and train a world-leading biopharmaceutical manufacturing workforce, fundamentally advancing U.S. competitiveness in this industry. NIIMBL seeks to share industry-wide considerations that could enable the biopharmaceutical industry to better deploy innovative technologies for manufacturing or continuous improvement. On May 23, 2019, NIIMBL held an Active Listening Meeting with representatives from 11 large biopharmaceutical manufacturing companies and U.S. Food and Drug Administration (FDA) stakeholders. The scope of the meeting was limited to topics of relevance to the Center for Drug Evaluation and Research (CDER) at the FDA (i.e., biotechnology products specifically).
2. Process Overview
The Active Listening Meeting format was piloted as a way to have a different kind of conversation between the regulated industry and the FDA, while reducing the risk of participation for both groups. For this pilot event, we sought to better understand the reasons behind the challenges in implementing new technologies in biopharmaceutical manufacturing.
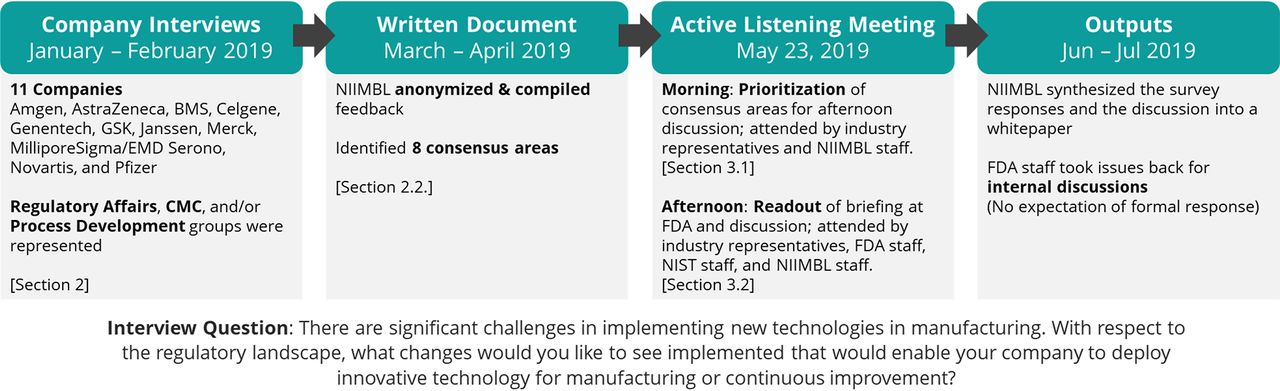
NIIMBL staff met by phone with individuals from Regulatory Affairs, Chemistry. Manufacturing, and Controls (CMC), and/or Process Development at 11 large global pharmaceutical manufacturing companies during the period of January–February 2019 (Figure 1). In scheduling the interviews, the companies were asked to consider the following questions as prework:

Process overview for Active Listening Meeting preparation and execution.
There are significant challenges in implementing new technologies in manufacturing. With respect to the regulatory landscape, what changes would you like to see implemented that would enable your company to deploy innovative technology for manufacturing or continuous improvement?
2.1. Guiding Questions for Discussion
As a prompt to help organizations think about responses, a number of guiding questions for discussion were suggested.
What is not working well today? Process, tools, and/or technology?
What do you need that you don’t have?
Is there sufficient awareness at all critical points within your organization about these challenges?
Is there any disconnect between what is perceived as the hurdle and the actual impediments?
What competencies, skills, experiences, and knowledge exist in staff/managers/suppliers?
What are the written or “unwritten” rules?
How much of a concern are post-approval changes?
Each organization was free to determine who would participate and what level of internal discussion and preparation was to happen in advance of the call. Each call began with an overview of the Active Listening Meeting process and the overall question about changes that could enable innovative technology in manufacturing. NIIMBL took notes on the interviews, anonymized the feedback, and identified areas of consensus. The same NIIMBL staff participated on all of the calls.
The 11 organizations that participated in this effort were: Amgen, AstraZeneca, BMS, Celgene, Genentech, GSK, Janssen, Merck, MilliporeSigma/EMD Serono, Novartis, and Pfizer. For the purposes of this publication, “industry” refers to the individuals who participated from these specific companies and is not a broad generalization beyond this group, unless stated as such.
Eight consensus areas were identified related to the above question: (1) interaction between the industry and the FDA, specifically related to transparency, collaboration, early interactions, and case studies; (2) changes to approved manufacturing processes, specifically approaches to comparability and post-approval changes; (3) operation in a global regulatory environment; (4) Consistency across the FDA; (5) guidance documents, with a particular focus on ICH Q12 (1); (6) manufacturing process development, specifically setting specifications and use of prior knowledge; (7) additional regulatory pathways or tools; and (8) education and training. The consensus areas and the number of interviews (out of 11) that touched upon a topic are summarized in Table I.
Consensus Area Summary
On May 23, 2019, stakeholders from these companies, the FDA, the National Institute of Standards and Technology (NIST), and NIIMBL met for an Active Listening Meeting. An industry-only session was held in the morning to align and prioritize discussion topics for the afternoon. The two consensus areas that were prioritized by industry for the afternoon discussion were: (1) interaction between the industry and the FDA, and (2) changes to approved manufacturing processes. An afternoon session was held with all participants during which NIIMBL presented the consensus topics and ideas shared by industry and gave all participants the opportunity to ask questions or comment, as appropriate. The FDA staff present at the meeting had the opportunity to internally discuss some or all of the areas identified as they deemed appropriate, with no expectation for a formal response.
This Active Listening Meeting was a pilot effort designed to create an alternate mechanism for feedback and conversation. NIIMBL looks forward to future efforts with different stakeholder groups on additional topics.
3. Pre-Meeting Findings
The authors organized and summarized the interview discussions. These findings were made available to all meeting participants.
3.1. General Reflections
It was clear that the industry has had a broad spectrum of experiences and perspectives in their interactions with FDA CDER. This diversity was evident both among companies and among functions within a given company. Although perspectives ranged from what could be deemed to be conservative to progressive, most companies fell somewhere in the middle. Companies that were more conservative were reluctant to introduce new technologies and preferred to use processes and techniques that had been successful in past regulatory filings, whereas more progressive companies incorporated newer technologies and strived to work with regulators toward shared understanding. Industry invests significant effort into considering when to implement new technology but are cautious about being the first to implement a new technology because of the risk to their application. Given the uncertainty of the interaction with the Agency around new technologies (and the global regulatory environment), there are relatively few business cases that justify the inclusion of new technology. Although there were several examples of positive collaborative interactions with the Agency, it was reflected that organizations feel powerless to question the FDA during an active submission. Despite the availability of a formal response procedure, this pathway was rarely employed by the companies interviewed because of the relationship between the industry and the Agency and because it would add additional risk to submissions. Industry approaches to FDA engagement ranged from some companies that deem "any interaction to be a Type C meeting" to others that consider some of the existing paths to interact with the Agency early (e.g., the Emerging Technologies Team) as a means to share and collaborate in learning. The basis for these different cultures among organizations is unclear. Each company culture is unique and is influenced by factors such as risk tolerance, organization size, product portfolio, leadership, history of interaction with regulators, and others.
Under the current system, there is rarely a business case for implementing new manufacturing technologies. Approaches to implementing new technologies were diverse, but a common sentiment was that there is rarely a business case to do so. There was no consensus on the best scenario to introduce a new manufacturing technology. Implementing a new technology in a Phase I investigational new drug (IND) may be viewed as lower risk; however, these products are many years from commercial approval and have a lower likelihood of success in the clinic. There was hesitation to use a new technology on an accelerated clinical program because speed is the most important consideration and new technologies pose a risk to timelines. There are also business risks associated with implementing new technologies post-commercialization. Therefore, there are few programs that represent a “sweet spot” for implementing new technologies.
Further, there is general aversion to being the first to deploy a new technology in a manufacturing process because of the perception that a sponsor may face overly burdensome hurdles during a regulatory filing. In general, regulatory risks, most commonly with respect to questions that can impact timelines, are perceived as real risks and are a barrier to implementation of new technologies. Perceived scientific risks, for example using new analytical technologies and detecting impurities in a product that has been on the market for a long time, can also be a barrier to implementation of new technologies. There is a gap between actual technical risk and perceived regulatory risk. It was not uncommon to hear about a process developed using new technologies, but a business decision being made to revert back to traditional technologies for the process at a larger scale and for formal submission based on a sponsor's internal perception of risk.
Although new technologies may offer some process improvements, those are weighed against business risks associated with speed to market. In this context, Regulatory Affairs departments may generally advocate for a more conservative approach with mature technologies rather than an approach with new technologies and business risks to the approval timeline. Mechanisms and approaches to address this situation would serve the industry (and patients) well.
3.2. Compiled Feedback from Industry Interviews
The number at the end of each heading represents the number of interviews in which a given topic was discussed. There were 11 total organizations interviewed.
3.2.1. Interaction between Industry and the FDA (11).
Individuals at the FDA often serve many different roles, which can include that of a product reviewer and that of a regulatory scientist. Survey participants felt that it would be helpful to have more engagement with the FDA in their role of regulatory scientists, using mechanisms that do not compromise their role as regulators.
Transparency (10). It was reflected that a better understanding of the Agency’s philosophy, context for inquiries, and opportunities for earlier and more informal engagement could improve the overall quality and speed of submissions.
Shared learnings for implementing new technologies—A mechanism by which the FDA could share common gaps in submissions and contribute to discussions regarding advanced technologies or strategies that have been successful would reduce industry-perceived risks. These could take place in the context of public workshops or in a blinded fashion through a consortium or other mechanism.
Context for reviewer questions—There is a desire to better understand the context in which a particular review question is generated. Precompetitive sharing in an open forum that allows discussion to build a shared understanding of the context for reviewers’ questions would be very helpful for both understanding and adequately addressing the question.
Benchmarking and data sharing—FDA staff have access to an abundance of data and it was understood that there are sensitivities in sharing data, even if it is anonymized appropriately. It was reflected that talks given by FDA staff at conferences, particularly those that use case studies, are extremely helpful. The most helpful learnings are gleaned from a statement of the problem, how the FDA viewed the problem, and how they approached solving the problem. However, the potential for public scrutiny of FDA presentations and the disconnect that occurs after the meeting are limitations to collaboration.
Volume of data shared in regulatory filings—There were questions around the right amount of information to share with regulatory authorities and a sense that there is a trend toward an ever-increasing volume of data, not all of which may be useful or necessary even from the sponsor perspective. Further clarity around what information is needed in a dossier versus what can be managed by internal quality management systems would be helpful.
Collaboration (10). There was a sentiment that both industry and the Agency would benefit from increased collaboration to facilitate industry taking more business risk in implementing new technologies. Public–private partnerships are a potential mechanism for such collaboration. One idea is for industry and the Agency to use a consortium such as NIIMBL to engage with scientists broadly in forums that allow the participation by the Agency as regulatory scientists (as opposed to regulators), on a NIIMBL platform process.
Platform processes—Platform processes run by consortia such as NIIMBL could also be used to generate comparisons between emerging technologies and traditional technologies. It could be a forum for technology-oriented and non-product-specific discussions about what data might be appropriate to include in a regulatory filing. This information could be made available in the form of peer-reviewed publications.
Public workshops—Another avenue to increase collaboration could be more FDA-driven public workshops on new technologies. These could provide an opportunity for early dialog between regulators and industry members, ideally with the shared goal of expediting emerging technology implementation to improve the manufacturing of biopharmaceuticals. Some example topics raised included: multi-attribute methods, real-time release, traditional stability studies (particularly use of accelerated degradation for prediction and modeling), continuous manufacturing, and rapid testing (microbial, sterility, mycoplasma, etc.).
Early interaction and the Emerging Technologies Team (ETT) (9). Feedback about the CDER Emerging Technologies Team (ETT) was positive; however, nearly all the experience reflected was with small molecules rather than biotechnology products. Positive aspects of the ETT include the willingness to openly engage in a dialog at the conceptual level and having a point of scientific engagement with the Agency outside of the context of the review of a particular submission. It was reflected that it is helpful to have an internal expert on a technology that can be the bridge to the individual reviewer or inspector, which helps build experience and improve comfort levels. Engagement with the ETT was often viewed as a formal FDA interaction and the amount of data required for the interaction perceived as prohibitive.
Early engagement—Although the ETT is a valuable and perhaps underutilized mechanism, there is industry desire for more early engagement outside of the context of a particular filing. These early interactions could take the form of a forum to ask questions, perform benchmarking, or to find out what general approaches have been successful in the past.
Informal interactions—It was stated that there was a general understanding that these informal interactions would complement, but would not replace the need for more formal approaches (e.g., to expedite regulatory pathways for post-approval changes). There is a desire for more options for how to provide information and data to the Agency for post-approval changes, driven by a desire to improve at the formal submission stage.
Case studies (9). Case studies, such as the A-Mab case study, provided a safe space for individuals to share their thought processes and to learn how others in the industry and at the Agency thought about different issues. Many viewed the outcomes of the A-Mab case study as valuable guiding principles for quality by design (QbD) and risk assessment and as important for establishing a common language and understanding.
However, for the A-Mab case study, there was a sense of a disconnect between the outcomes published and what happened in practice with respect to QbD. Future endeavors would benefit from a mindful approach to minimizing the disparity between expectations and reality and tempering expectations.
Future case studies could generate non-product-specific dialog about new technologies between industry and the Agency. Pitfalls could be avoided with clear communication throughout the exercise, such as by choosing a topic that addresses a relative and immediate need, and by outlining clear goals and objectives of the exercise. An example topic includes a mock early-stage filing that uses new technologies for a hypothetical molecule that implements ICH Q12 (1) concepts in a filing, specifically analytical comparability using a new technology. Another example would be “A-Mab 2.0”, which would use the same hypothetical molecule but focus on a process based entirely on new manufacturing technologies.
3.2.2. Changes to Approved Manufacturing Processes (11).
Approaches to comparability (10). Demonstrating comparability often requires clinical data, and collecting this additional data can be expensive and time consuming. In contrast, it was also reflected that the expectations for analytical vs. in vivo vs. clinical data were very clear and these decisions were managed internally to be consistent with Agency expectations. Implementing new technology and deciding the best path to establishing comparability often includes the business risk of potentially needing to collect more clinical data.
Implementing a new technology might improve product quality in known ways, but a change to product quality is still a change to the product and would require clinical data.
A question was raised: Is comparability an adequate tool to bridge an existing technology with a new one? It may not be possible to truly compare Old Technology A to New Technology B, but rather a new assessment of the new technology might be more appropriate. Community-wide discussion of frameworks for this decision-making would support the industry.
Post-approval changes (10). There is a strong desire from industry to have a conversation about how to implement changes to processes more efficiently. In general, the FDA was viewed as reasonable in their approach to post-approval changes. There is an aversion to making changes that could have an unanticipated change in patients and a disruption in brand.
Topics that were raised in multiple interviews included:
Knowing when analytical data is "good enough" vs. needing clinical data.
Ways to accelerate the review cycle timeline.
Using knowledge from past experiences in current conversations.
Disconnect between company risk assessment and Agency risk assessment.
Operating in a global regulatory environment, with the potential need to maintain multiple processes and supply chains to meet different Health Authority requirements.
Desire for greater reliance on internal quality systems to manage post-approval changes, especially minor changes.
Differentiating between magnitude of process change and magnitude of product change.
Suggestion that the potential for more interaction throughout the post-approval process could allow organizations more insight into the process and could enable the ability to respond to questions or gather additional data on an accelerated timeline if necessary.
3.2.3. Operating in a Global Regulatory Environment (11).
All companies operate in a global regulatory environment and every company that participated reflected that this is a huge challenge. Each health authority has different timeline requirements, requires different discussions, and requires different amounts and types of information. There was recognition that this is not an FDA-specific nor CDER-specific problem.
One common sentiment was that it is not a practical business decision to run and maintain a supply for two different processes for different groups of countries, and this has a significant impact on whether or not to pursue process changes.
There is an appreciation that the FDA engages with the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), the World Health Organization (WHO), and a number of different international forums. Although the burden of harmonization is not on the FDA, companies were hopeful that the FDA has the opportunity to influence and take a leadership role in the global regulatory environment. There is a desire for alignment on the science and the risks associated with the science. If the top markets were aligned in their interpretation of ICH guidance and in their requirements, that would alleviate a huge burden. Specific markets mentioned were the United States, the European Union, Japan, Canada, Brazil, and China. In addition, the WHO could be leveraged to reach alignment across a larger number of countries with their own internal mechanisms.
3.2.4. Consistency across the FDA (8).
There is a desire for consistency across the FDA, which includes between individuals (e.g. different reviewers or inspectors), between the review and the inspection process, and between leadership (“the podium”) and reviewers/inspectors.
Between individuals (reviewers or inspectors) (7). There was acknowledgment that each person is an individual, but there were notable differences between seasoned and newer members of the Agency staff. Experience and comfort level could drive these differences and could, in part, be addressed by education and training. An additional suggestion was an internal Agency quality control process that, for example, pairs up reviewers for training.
Between leadership/policy and reviewers/inspectors (3). It appears that the Agency as a whole, and particularly leadership and policymakers, want to move technology forward and embrace innovative approaches and novel strategies. However, the implementation of these approaches at the review and inspection level is often considerably more conservative than the messages received from the podium. The perception is that there is less comfort with new technologies and approaches, and the associated real or perceived risks, at the individual level. Education and training, increased early interaction, and perhaps other mechanisms could be developed to improve comfort levels of individual reviewers, in alignment with the goals set out by leadership in support of technology advancement and innovation.
The disparity between leadership and day-to-day processes can also be seen at scientific conferences. Leadership from the Agency often encourage people to engage in new technologies as appropriate, but the reviewers tend to take a conservative approach to filings. Encouraging and improving scientific sharing and collaboration could improve consistency between messages heard in public forums and experienced in day-to-day practice.
Between reviewers and inspectors (3). There was a sense that the line between review and inspection may be blurred, even though the objectives of these functions are different. It was acknowledged that inspectors are generally diligent about knowing where questions cross the line from what is directly in front of them to something that needs to go back to the reviewer. It was reflected that having a product reviewer on site can result in a different (and more positive) interaction and there was encouragement for CDER to maintain this practice. Additional education and training are also potential mechanisms to improve consistency and understanding of boundaries by all parties.
3.2.5. Guidance Documents (8).
Companies are constantly seeking updated and new guidance documents from the Agency. A revisit of “Changes to an Approved Application for Specified Biotechnology and Specified Synthetic Biological Products” (2) would be welcome. It was also reflected that the principles in guidance documents might still apply, but the examples provided might be out of date (specifically mentioned was ICH Q5E). It was reflected that the industry would benefit from faster release of guidance, especially related new technologies and approaches, acknowledging that there is a process to issue such guidance. There was also a desire for less formal input from the agency in the form of case studies, best practices, or other learnings that could be shared to identify gaps in submissions or successful approaches to improve the quality of submissions overall.
ICH Q12 (3). The ICH has developed a draft guidance to harmonize post-approval CMC changes, ICH Q12 (1). As of December 2019, the guideline entitled “Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management” had been adopted (Step 4 of the formal adoption procedure) and is ready for regulatory implementation in ICH regions. Companies are hopeful that the new ICH Q12 guidelines will shape the future regulatory environment. The document has the potential, if successful, to deliver on streamlining the review process for commercial products, which is estimated to currently take 3–5 years.
There is concern, however, about how the different regulatory authorities might interpret ICH Q12, related to operating in a global regulatory environment. This could be a great opportunity for the FDA to take a leadership role in alignment on the concepts outlined in ICH Q12.
There was a suggestion that a public–private partnership could facilitate a way to work through the details of what this guidance might look like in practice through a case study.
3.2.6. Manufacturing Process Development (7).
There were several discussions around barriers to implementing new technologies centered around manufacturing process development.
Specification setting (4). Process specifications are currently based on small numbers of clinical lots and represent a relatively homogenous data set, whereas a pool of manufacturing data can be quite different as molecules are often manufactured at multiple facilities. Companies desire operational and scale-up flexibility to match any needed lot size. It would be helpful for industry and the Agency to collaboratively examine commonly measured attributes in terms of what they do and do not represent for the safety and efficacy of the product.
One potential idea was that a product could gain preliminary approval with an initial (and small) number of clinical batches, and then come back at a later date with more manufacturing data to set appropriate specifications. Another idea centered around using prior knowledge in regulatory filings to set specifications broader than those set by clinical experience alone.
Using prior knowledge in regulatory filings (6). A wealth of platform-based data, for both processes and molecules, exist within industry. Use of platform data to support regulatory filings could expedite and simplify the regulatory submission and review process. There were questions around how information used in one application could be cross referenced, if similar and applicable, to another product at a different site, to leverage prior knowledge without putting in an independent submission. A broader scientific conversation could be held around what science, technology, or prior art can be deployed to widen specifications.
3.2.7. Additional Regulatory Pathways/Tools (7).
There were a number of ideas for additional regulatory pathways or tools to lower the business risk and accelerate the timeline for implementing emerging technologies in biopharmaceutical manufacturing (with the goal of accelerating patient access to medicines while still assuring safety, efficacy, and supply).
Drug master file equivalent for new technologies—This type of tool could be used for technologies referenced by multiple organizations or used by the same organization for a multi-site or a multi-product submission (e.g., platform technologies; see Using prior knowledge in regulatory filings).
Breakthrough/accelerated/priority/fast-track technology designation—This tool would provide an expedited pathway, ideally at the post-approval stage. Such a tool could provide incentive within a company and reduce the timeline risk for using a new technology in a program with high value.
More interaction during submissions and approvals—Particularly with respect to post-approval changes, more engagement throughout the process could enable a company to respond more quickly.
Vendor interaction—A public–private partnership, like NIIMBL, could be used to enable direct vendor engagement with the Agency to gain scientific understanding of new technologies, assays, and approaches.
3.2.8. Education and Training (5).
It was reflected that both industry and the FDA require a highly trained staff. The industry as a whole could benefit from more applied education, such as bioinformatics or applied process engineering. The ability to adopt new technologies (from both sponsors as well as regulators) relies on scientific understanding of new technologies and better education drives a "comfort level'. Additionally, all parties could benefit from better education on risk-based approaches.
4. Meeting Summary
NIIMBL met in the morning with industry only to discuss and prioritize topics for the afternoon discussion. All stakeholders, including industry, FDA, NIST, and NIIMBL met in the afternoon.
4.1. Morning Session—Discussion with Industry Representatives Only
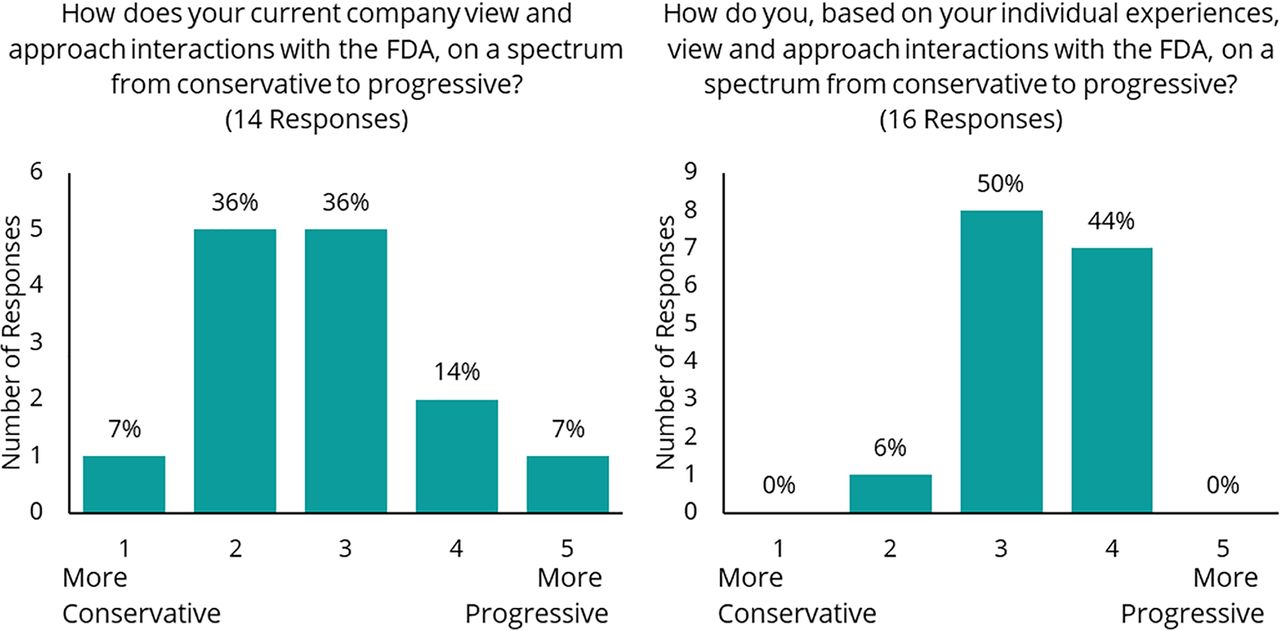
A morning session was held with industry representatives and NIIMBL staff to elaborate on experiences shared in the prework interviews and to prioritize discussion areas for the afternoon. An anonymous, real-time polling tool was used to poll individuals about their perception of interactions with the FDA and their priorities. When asked to rate their current company’s view and approach to interactions with the FDA, on a scale of 1 (conservative) to 5 (progressive), the average response was a 2.8 (14 responses; Figure 2 left). When asked to rate, based on individual experiences, their view and approach to interactions with the FDA on the same 1 to 5 scale, the average response was a 3.4 (16 responses; Figure 2 right). There was discussion around potential reasons for the differences between the overall company perspective and the individual perspective. It was postulated that individual interactions are able to be less formal and more science-based, whereas an interaction on behalf of a company is, by necessity, more formal.

Anonymous, real-time poll results from morning Industry-only session.
The poll was also used to identify the priorities for the Active Listening Meeting and discussion with the FDA. The top two priority topics were (1) interaction between the industry and the FDA and (2) changes to approved manufacturing processes. When asked what were the main messages that individuals would like to convey in the afternoon session (free text), many responses (9/17) shared the sentiment that more opportunities for informal discussions about new manufacturing technologies could improve the implementation of new manufacturing technologies. There was an appreciation for the FDA and a desire to collaborate and work together to solve some of the industry’s biggest challenges.
4.1.1. Interaction between the Industry and the FDA.
On the topic of interaction between the industry and the FDA, there was a recognition that there is a large diversity in approaches both within the FDA and within a given company. There was a general feeling that the ability to talk to a reviewer has decreased over the past decade, and that CDER reviewers are more formal than reviewers from other centers. There was a desire to be able to have a conversation based around data, which may or may not be complete, toward understanding if a particular approach might be promising. There was broad agreement among industry representatives that fear of receiving a final, formal “no” from the FDA can be a barrier. For the culture between industry and the FDA to change and to improve the nature of informal interactions, there should be a reduction in fear that everything the FDA says will be taken as formal guidance, and the industry must "stop taking everything" the FDA says as formal guidance.
4.1.2. Changes to Approved Manufacturing Processes.
There was significant discussion around post-approval changes to manufacturing processes, and particularly the business case around these changes. There was discussion about the different risks to implementing new technologies at different stages, including risks to timelines and the complexities of managing changes in a global environment. There was discussion about how best to bridge old and new technologies. There was a consensus that it is important to be driven by science; however, technologies with good science may still experience a delay in approval because of unfamiliarity. There was a desire to better understand successful approaches taken by other organizations but a simultaneous understanding of the need to protect a competitive edge or proprietary information. Industry representatives shared that there have been examples of new technologies being used to successfully widen specifications, but this requires a risk on the part of the company. There is a significant widespread aversion, resulting in internal barriers, to the possibility of needing clinical data to implement a change to a manufacturing process.
4.1.3. Consistency across the FDA.
A final significant area of discussion was around the context and consistency of reviewer questions. Representatives suggested that there could be opportunities for training or exchange of knowledge. Coupled with the desire for more interaction, there may be a way for the Agency to evaluate and prioritize efforts that promote additional feedback in the areas where they want to see companies invest and grow.
4.2. Afternoon Session—Active Listening Meeting with Industry Representatives, FDA, and NIST
The attendees for the afternoon session included all the industry representatives from the morning, representatives from the FDA (primarily from CDER but including individuals from CBER and CDRH as well), NIST staff, and NIIMBL staff. NIIMBL presented the key findings, with a focus on the priority areas identified by industry in the morning session.
4.2.1. Business Case for Adoption of New Technologies.
The afternoon discussion began around the components of the business case for the adoption of new technologies. It was agreed that to say there is “no” business case is an oversimplification. There may be a business case, but it is perceived as not strong enough to overcome hurdles, whether real or perceived. There are separate challenges for implementing new technologies in early-stage projects, which have a lower likelihood of making it to market, as well as in late-stage projects where timelines are especially important and which also have the added challenges of a global regulatory environment. The nature of the global regulatory environment also limits opportunities to implement new technologies post approval. From the company perspective, there is a desire to implement a new technology for multiple products at the same time. Companies are constantly seeking confidence that the path to approval with a new technology will not be more difficult or require significantly more time than using a more well-known technology. Time delays may be unacceptable to certain programs, and the uncertainty of the timeline of the path to regulatory approval for processes using new technologies is often enough to keep a company from implementing the new technology.
There was discussion on the differences between cost and value, as well as risk and opportunity. There was also discussion on whether the cultural drivers for an overall conservative approach are because of significant past high-impact events or the “sum of fears.”
There was agreement that Contract Manufacturing Organizations (CMOs) will have an even harder time adopting new technologies until the industry reaches a “consensus” because of the need to convince multiple companies to buy their manufacturing capacity.
4.2.2. Interaction between Industry and the FDA.
There was much discussion about the types of interactions, and the industry representatives' desire for more informal interaction. Informal feedback could help organizations internally de-risk implementing a new technology; however, culture changes are required to be able to have these types of interactions in a productive way. It was agreed that there appears to be a direct relationship between the specificity of a question and the formality of the response, with more specific questions requiring more formal interactions. Further, operating within complex organizations is a challenge for both the industry and the Agency, as different individuals and groups interface at different points in the process.
A safer space for interaction could center around scientific considerations such as risks, issues to be resolved, and parameters to be considered, and less about specific issues. Workshops can be a useful venue to have precompetitive discussions, but every organization has a different definition of when a conversation strays into competitive territory. A public comparability database could be another venue for precompetitive sharing of scientific data. The community could also benefit from more discussion of failures as well as successes.
Case studies. There was significant discussion around case studies as a tool to facilitate interaction and to establish a common vocabulary. A-Mab (3) was a significant effort and was widely viewed as a positive experience, but was published in 2008. There are other ongoing efforts such as A-Vax (4) and A-Gene (A-Gene is an ongoing effort supported by the Alliance for Regenerative Medicine and NIIMBL). There was enthusiasm from both the industry and the Agency for potential future case studies as resources allow.
Collaboration and shared learnings. There was agreement between the industry representatives and the Agency that shared learnings can make everyone more effective in their roles; however, there was no good suggestion as to how those might be collected and shared. The FDA does not “own the information” and individual organizations do not share regulatory successes at open meetings on a regular basis.
3.2.3. Changes to Approved Manufacturing Processes.
There was discussion around the comparability protocol as a tool for post-approval process changes. One suggestion was a workshop around a novel and theoretical manufacturing product or process to identify what the risks are and are not, in the context of implementing the technology in a post-approval context.
3.2.4. Consistency across the FDA.
There was a significant discussion around consistency of reviewer questions during information requests. There is a desire from industry for more context about why a particular question might be asked. It was reflected in multiple interviews that there were experiences of getting no questions for a given technology in one process, and many questions around that same technology in a similar process, which creates risk. It’s important to know when there are significant differences in lines of questioning and for all parties to understand when a certain context is important. There are inefficiencies on both sides, with information requests that don't clearly articulate the reason for the question, and not understanding the question and not fully responding, which can lead to many rounds of written back-and-forth that can add significant delays. These delays can be internal to companies as well. There was a discussion about the value of identifying the missing information, providing a reason for why it is needed, relating the reason to safety and/or efficacy, and making the specific request.
Every company and every reviewer have some experience that drives how they approach different situations and therefore there are challenges around individuals with many years of experience as well as challenges with more junior staff. This is an opportunity where NIIMBL could support the community by brokering the collection and sharing of relevant and appropriate experiences.
When discussing new regulatory tools, a key consideration is the benefit to public health. For example, if implementing a new manufacturing technology can help relieve a drug shortage, there is a clearer benefit to public health, and this could generate additional conversations. There are other mechanisms, such as legislation, that could help incentivize innovation and support repatriation of biopharmaceutical manufacturing to lower the risk to the quality, consistency, and availability of biopharmaceuticals.
5. Conclusions and Next Steps
The Active Listening Meeting mechanism employed here has not previously been tested and has led to meaningful exchange of ideas and outcomes. NIIMBL’s external evaluators collected anonymous feedback via a short survey after the event. Of the 12 individuals that responded (with not all individuals answering every question), the mean satisfaction was 3.73/5.00 (between “satisfied” and “quite satisfied”) and participants agreed that the workshop was a good use of time (4.27/5.00, between “agree” and “strongly agree”). Further, 90% of respondents indicated interest in attending a future workshop (10% maybe). Additional qualitative feedback indicated that participants were satisfied with the attendance of key stakeholders from the FDA and from industry and appreciated the format and the frank discussion at the meeting.
There are many opportunities to follow up on the learnings from this effort. This Active Listening Meeting, brokered by NIIMBL, represents a new method to share information between the industry and the Agency in an informal way. A natural set of next steps involves additional Active Listening Meetings focused on different topics. Potential areas for discussion include: a more focused discussion on one section of this publication, similar discussion around different product types (e.g., vaccines or cell and gene therapies), or interviews with different stakeholder groups such as small companies or academic institutions. Another suggestion for follow up was to develop a framework around precompetitive manufacturing, to identify safe areas for discussion among organizations and the Agency, and to draw a line for where discussions may venture into the competitive space. There was general enthusiasm for case studies that brought the industry and the Agency together. Additionally, collaborative benchmarking studies and data sharing would benefit the community broadly.
Conflict of Interest Statement
The authors declare no conflicts of interest.
Acknowledgements
We are extremely grateful for the time and input from all of the participants. This includes individuals from Amgen, AstraZeneca, BMS, Celgene, Genentech, GSK, Janssen, Merck, MilliporeSigma/EMD Serono, Novartis, and Pfizer. It also includes the input and engagement with FDA staff from OCS, CDER, and CBER. JM and KHL are supported in part by award 70NANB17H002 from the U.S. Department of Commerce, National Institute of Standards and Technology.
- © PDA, Inc. 2020




{kind=link}
{kind=link}