
Abstract
Pre-use/post-sterilization integrity testing (PUPSIT) has been a widely debated topic for the last several years. To a large extent, the debate is because of the fact that scientific data were not available to provide additional clarity that could inform appropriate risk-based judgements and commensurate actions. To gain clarity, the Parenteral Drug Association (PDA) and BioPhorum Organizations Group (BioPhorum) formed the Sterile Filtration Quality Risk Management (SFQRM) consortium in late 2017. The consortium goals have been to fill existing gaps in scientific data as adequately as possible with studies and industry guidance that would provide professionals and license holders with the ability to make informed decisions about appropriate risk-management strategies. This paper is one in a series of publications that are the result of the collaboration, and these should be considered together and viewed holistically in order to determine the best course of action with regard to PUPSIT. In total, the four papers cover the following areas: (1) data mining to determine the influence of fluid properties on integrity test values, (2) filter masking studies and results (this publication), (3) risk assessment and management from filter production to end use, and (4) points to consider in the best practice of the use of PUPSIT. In total, 25 manufacturers and filter suppliers have contributed to the work of the Consortium, deploying their filtration experts and pooling their collective knowledge and applied science experience to address these questions. This effort has also been supported by many independent experts currently available who have contributed to and driven the Filtration Interest Group in the PDA for many years. Both PDA and BioPhorum have prioritized this program and combined their approaches to deliver this comprehensive body of work. We hope that collectively the publications aid decision making and create greater certainty and confidence and above all alignment between suppliers, manufacturers, and regulators alike on these important questions.
Introduction
Sterilizing-grade filtration has been reliably used for decades and increasingly so, as more medicinal products are biologics, which prohibits the use of heat sterilization. The reliability of this critical aseptic processing step improved with new filter materials, designs, and the adoption of single-use filtration technology to create a functionally closed-unit operation. Moreover, process validation activities, which evaluate the performance of a sterilizing-grade filter under process conditions, support the reassurance that the filter performs as specified and a sterile filtrate is obtained (1⇓–3). The validated state of the filtration process can be verified by confirming that the sterilizing-grade membrane filter is integral by performing nondestructive integrity tests, including the bubble point, diffusive flow, or pressure decay tests. The integrity test can be performed pre-use/pre-sterilization, pre-use/post-sterilization (PUPSIT) and post-use (“use” meaning the actual filtration process). The post-use test is a regulatory requirement.
The integrity test of a sterilizing-grade filter has to be and most commonly is performed after the filtration process (post-use). Some filter users test the integrity before the filtration process and before the filter is sterilized (pre-use). Less common is the integrity test performed after the sterilization of the filter before use (pre-use/post-sterilization or PUPSIT), as such tests would require downstream, filtrate-side manipulation generating a potential process risk. Therefore, most regulatory authorities request the test of the integrity of a sterilizing-grade filter be performed after the filtration, post-use, but only recommend an integrity test pre-use, without specifying whether it should be performed pre- or post-sterilization (2⇓⇓–5).
However, since 1998, the EU Guidelines to Good Manufacturing Practice: Medicinal Products for Human and Veterinary Use, Annex 1 (Manufacture of Sterile Medicinal Products) or “Annex 1” has contained the requirement for verifying the integrity of a sterilizing-grade filter before use and after its sterilization, by stating “The integrity of the sterilised filter should be verified before use and should be confirmed immediately after use by an appropriate method such as bubble point, diffusive flow or pressure hold test....” The requirement remained in the 2008 revision and in the 2017 draft revision to Annex 1. Although not enforced for years, sporadic demands for PUPSIT started to occur since 2007. To determine the motivation of the enforcement of the pre-use/post-sterilization integrity test, multiple meetings and discussions were held. It appears that the main argument for the need to perform a PUPSIT is the possibility that a flawed pre-use sterilizing-grade filter could pass the post-use test, as the flaw could be masked by the separated contaminants from the fluid stream.
The foundation of the enforcement was a published EU GMP Guide Annexes Q&A (7) of the same year, which states:
“The filter-sterilisation process may be physically stressful for the filter. For example, high temperatures during the process may cause the filter to distort, potentially leading to fluid pathways that allow the passage of particles greater than 0.2 µm in size. The performance of a filter can improve with use, as particles begin to block individual pathways and remove larger pathways that smaller particles could successfully navigate. For these reasons, filters should be tested both before use but after sterilisation and again after use.”
Based on the language in the 2007 Q&A statement and also statements made by European regulators, it became clear that the flawed filter masking is a major reason why PUPSIT is now being enforced. Most of the discussions and concerns surrounding filter masking that have been reported have been based on conjecture or anecdotal accounts, rather than published scientific studies and analysis and there has been no direct, measurable scientific evidence that this ever happened.
Recognizing this lack of data, the Parenteral Drug Association (PDA) and BioPhorum Organizations Group (BioPhorum) created a joint task force to determine whether or not masking of sterilizing-grade filter flaws is possible. With the invaluable support of filter manufacturers and the PDA Training and Research Institute (TRI), masking trials were performed, which will be discussed within this paper.
Reviewing the Sterilizing-Grade Filtration Step
It is worthwhile to note that sterilizing-grade filters undergo a high degree of inspection by the filter manufacturers and the end users alike. Quality checks are performed throughout the filter manufacturing process beginning with the membrane, which is evaluated for bacterial retention, integrity, and other critical quality attributes. The PDA/BioPhorum risk-assessment task force team detailed all the evaluations in their paper “Risk assessment and management from filter production to end-use.”
During filter manufacturing, all sterilizing-grade filters are required to pass an integrity test during the filter manufacturing process, as it is one of the release criteria. When a filter fails the internal integrity test, it will not be released, and the failures are investigated to determine the root cause. This in-process integrity test evaluates the integrity of the membrane as well as the integrity of the seam and end-capping process. When flaws are found, the membrane lot or filter element is rejected, and a root cause analysis triggered. All tests have the ability to detect defects with accuracy, as it is of the utmost importance for the filter supplier to deliver highest quality products.
Furthermore, the packing and transportation of these filters is examined and validated to avoid filter damage during shipment to the end user (8). The end user also has various acceptance controls to avoid any potentially damaged filters from entering the production process, including visual inspection, proper storage, and handling criteria. Personnel are trained on procedures that are established to instruct on handling, set up, and installation to avoid any damage at the use point. All in all, terminal sterilizing-grade filters are handled with care as all parties involved recognize the criticality of this unit operation for product quality and patient safety.
Sterilizing-grade filtration systems are required to be sterilized before use; either presterilized as part of a single-use assembly or sterilized in-situ or by autoclaving after connection into the process or filter housing. The presterilization process, commonly performed by gamma irradiation, is validated by the supplier of the single-use assembly and long-term validation studies have shown that filter systems are not at risk to be damaged by this process. However, when the membrane filter is installed into a stainless-steel filter housing and in-situ steam sterilized, filters may experience higher thermal and mechanical stresses, as mentioned in the Supplementary Requirements—Annex 1 Manufacture of Sterile Medicinal Products Q&A document (7). For that reason, steam sterilization qualification is a standard industry practice, not only to determine the sterilization efficacy, but also to establish whether the sterilization process stays within the maximum allowable operating parameters given by the filter manufacturer to avoid any sterilization damage. If the qualification has not been performed, performed poorly, or the end user has not been trained to perform a steam sterilization process, the filter could be damaged. Such damage can cause a flaw (commonly, gross failures) that may allow microbial penetration to the filtrate side. For this reason, post-use integrity testing is mandatory as it allows the detection of such flaw.
Aside from the sterilization process of the membrane filter, end users are required to review the terminal sterilizing-grade filtration step, as there are requirements that influence the risk assessment of the integrity test timing and needs. The position of the terminal filter is required to be as close to the filling line as possible (6), which means that the filter should have enough capacity to not block (plug) and does not require to be exchanged during the filtration/filling run. To determine the appropriate sterilizing-grade filter size or the combination of a prefilter/membrane filter, filterability trials are commonly performed with the actual product under processing conditions. These trials allow accurate sizing of the needed effective membrane filtration area to filter the specified batch volume in a desired time frame. However, to ensure a sterilizing-grade filter exchange will not be necessary during the batch filtration, end users customarily permit much less than 50% blocking of the filter element. Other end users utilize a protective prefilter in front of the sterilizing-grade membrane filter or the use of redundant sterile filtration. This conservative filter sizing approach ensures that the final sterilizing-grade filter should never experience a high load of foulants or blocking agents. In conclusion, adequate effort is taken to ensure that the filtration runs uninterrupted.
Need for Masking Studies
The hypothesis that an enlarged pore structure or minor flaw within a sterilizing-grade filter will be masked by fluid foulants so that the post-use integrity test cannot detect it has only been anecdotally reported. No scientific evidence has been available. Moreover, experiences by filter manufacturers having performed thousands of bacteria challenge tests, subjecting sterilizing filter units to 107 organisms per square centimeter, which represents a high contamination level in the feed stream (9), have not shown masking effects that could cover a flaw. This also has been verified by the PDA/BioPhorum task force data mining and the results will be published as “Datamining to determine the influence of fluid properties on the integrity test values.” Typically, the filter integrity is tested before and after the bacteria challenge test. Reviewed results showed that filters that failed the integrity test before being subjected to the high bioburden challenge test have also failed the integrity test post challenge. Additionally, comprehensive literature searches and discussions with end users across the pharmaceutical industry could not verify the anecdotal reports voiced or their source.
As scientific data to show whether masking of a filter membrane flaw can occur were not available (a key argument as to whether PUPSIT should be performed or not), masking studies were needed. To this effect, four major filter manufacturers, Meissner, MilliporeSigma, Pall and Sartorius Stedim, agreed to jointly support this project.
Activities Description and Reasoning
Test Protocol
To run the test trials in a synchronized manner, a test protocol was established (See Appendix). This test protocol had multiple reviews by the filter manufacturers, the task force members, and regulatory authorities. The test protocol described the test procedure, materials, and the foulant to be used in the study. The foulant used was Ovaltine, a proteinaceous malt, cocoa extract, at specific concentrations, which has been found to mimic biologic solutions exceptionally well. The differential pressure used to run the test fluid through the filter was held at a constant 10 psi, as this has been determined to be a typical pressure used in the final filtration applications. Once the protocol was approved by all parties, the Phase 1 test trials were executed in the PDA TRI.
Phase 1.
For the Phase 1 masking trial work, the filter manufacturers collected in total 24 × 10 inch filter cartridges of various membrane materials and configurations, four of those had a 0.4 µm pore size and 20 had a 0.2 µm pore size. The filters tested with automated integrity test machines were marginally nonintegral, meaning the integrity test values were close to the integrity test limits and not catastrophic failures (gross damage to pores). It should be noted that most nonintegral filters have catastrophic failures and that marginal filter failures are extremely rare. In this trial, a worst-case foulant concentration of 24 g/L was used and the blocking level of the filters was >90% (i.e., postexposure flow was <10% of the initial flow). The typical foulant concentration used to mimic biologic solutions is 0.8 g/L and as mentioned in practice the blocking level is <50%.
Phase 2.
For Phase 2 masking trial work, 0.2 µm, 47 mm discs of the same membrane materials and combinations as those used in Phase 1 were used; these were deliberately flawed by laser drilling a 10 µm hole. Based on the results obtained during Phase 1 testing, it was decided that the additional testing with a defined flaw would be able to provide a more robust dataset regarding the potential masking of minor flaws during filtration. In this trial, the foulant concentrations of 24 g/L and 0.8 g/L were used at a blocking level of 25%, 50%, and 75%. Once again, the 24 g/L was used to see the effect of a worst-case foulant solution and the 0.8 g/L solution was used to experience a more realistic case.
Result Reporting and Discussions
Phase 1
As mentioned, in Phase 1 the task force team jointly decided to use a worst-case fouling scenario for sterilizing-grade filters. The goal was to determine whether or not it is at all possible to mask a filter flaw with a plugging solution. From 24 filters tested, two that failed the pre-use test passed the post-use bubble point integrity test, meaning the flaw was masked (Table I).
Phase 1 Test Results: Pre- and Post-Use Integrity Test Results of 10” Filter Elements Subjected to a Foulant Concentration of 24 g/L and Blockage of >90%
This result finally represents scientific evidence that a flawed filter can be masked under extreme circumstances. The test team was encouraged by the results, as it created a basis of filter fouling, which can be compared to realistic processing conditions and therefore the trial work moved forward to mimic the realistic circumstances in Phase 2.
To support the statement of worst-case foulant and blocking scenario, Table II compares the typically found processing conditions to the test conditions used.
Phase 1 Test Condition Versus Typical Conditions in Biologic Applications
Phase 2
In the initial Phase 2 trials, the worst-case foulant concentration of 24 g/L was used to determine its behavior with lower blocking levels and also to see whether additional trials showed a similar rapid blocking behavior. In addition, a 0.8 g/L foulant solution was employed to more closely mimic typical biologics solutions. Both foulants were used at blocking levels of 25%, 50%, and 75%.
The work with the 47 mm discs was challenging in regard to the blocking level, as the filter surface area is very small. This was especially so for the 24 g/L concentration, which fouled the filters immediately in most instances. This did not allow the integrity tests to be performed, as the membrane discs could not be sufficiently flushed with the wetting fluid volume specified. Therefore, the 24 g/L data set is small. In three test instances automated integrity test machines were deployed and in one instance the manual bubble point test was used.
Table III shows the results of the 0.8 g/L foulant concentration tested with automated integrity test systems. The measurements with the test instruments often showed “abort” results, as the test instrument detected an excessive flow owing to the laser inflicted flaw. Table IV shows the results of the 0.8 g/L foulant concentration measured with the manual bubble point and the measured blocking levels in ascending order. With the use of a manual bubble point test, the actual bubble point was measured without an aborted result. For this reason, the two test results were split into two tables.
Phase 2 Test Results: Automated Pre- and Post-Use Integrity Test Results of 47 mm Disc Filters Subjected to a Foulant Concentration of 0.8 g/L and Blockage of 25%, 50% and 75%
Phase 2 Test Results: Manual Pre- and Post-Use Integrity Test Results of 47 mm Disc Filters Subjected to a Foulant Concentration of 0.8 g/L and Various Blockage Levels up to 99%
As shown in Table III, all filters failed the post-use test. As shown in Table IV, at a blocking level of 81% and 97%, two of the 27 filters passed the post-use test with the 0.8 g/L foulant. None of the filters passed the post-use test at the 25%, 50%, or 75% blocking level and 16 of 18 filters failed the post-use test at a blocking level >80%.
As mentioned, the data set for the 24 g/L foulant concentration was much smaller, as the 47 mm discs fully plugged in some cases and post-use wetting was impossible owing to the blocked membrane. Very few data points were established. Certain filter membranes contained a prefilter layer with a larger pore membrane in front of the 0.2 µm membrane, which may have reduced the fouling of the 0.2 µm membrane by the 24 g/L solution. Once again, some of the membranes were tested with an automated integrity test machine and others with the manual bubble point test. All test results are listed in Table V.
Phase 2 Test Results: Automated and Manual Pre- and Post-Use Integrity Test Results of 47 mm Disc Filters Subjected to a Foulant Concentration of 24 g/L and Blockage of 25%, 50%, 75% and up to 93%
Table V shows the few results that were obtained during testing. The data show that the integrity test instruments, as well as the manual test method, detected the flawed filters post-use even at a blocking level of 83% and 93%.
Results Conclusion
The tests performed created the first scientific data set on the impact of filter fouling or blocking on the post-use integrity test by multiple filter manufacturers, which shows that the use of PUPSIT is dependent on the application, especially the fluid properties. When the fluid is a high-foulant fluid, which blocks filters quickly, the possibility of masking a filter flaw is elevated. A low-foulant fluid with a lower blocking level does not present such a risk. As a result, it can be concluded that PUPSIT may be required for solutions that have an abnormally high blockage level. Knowing if the fluid to be filtered is a high- or low-fouling fluid allows the end user to conduct a risk assessment to determine the needed sterilizing filtration integrity test process for their specific application. Running filterability tests to size the sterilizing filter in a specific application creates the knowledge of the fluid/filter behavior, and as a result provides the information required (blocking level) to assess whether performing PUPSIT would be required to ensure flawed filters are identified. Filtration procedures require information about the process and fouling propensities of the fluid stream, rather than a blanket enforcement of test procedures that may bring their own risk to the process.
As described, the terminal sterilizing-grade filtration process does not allow significant blockage of the filter. For that reason, the majority of end users allow only a blockage level at which the process can be reproducibly controlled, generally <50%. Furthermore, biologic solutions are typically the higher foulant fluids experienced within the industry and are typically mimicked in filter test trials by the use of 0.8 g/L concentration Ovaltine or other similar solutions.
To determine whether there is any real risk of masking flawed filters such that the post-use integrity test would not detect the flawed filter, the test team decided to use a worst-case scenario in excess of a realistic case (Table II). The foulant concentration was 30 times higher than the normal 0.8 g/L mimic foulant fluid, at 24 g/L, and the blockage level was >90%. In this case, two of the twenty-four 10 inch pleated elements were found to be masked and passed the post-use integrity test.
To build a more robust data set that is closer to a more realistic biologics fluid and determine the critical process condition that may result in masking, additional trials were performed with different blocking levels and two different foulant concentrations, 0.8 g/L (realistic case) and 24 g/L (worst case). As it has been challenging to collect flawed pleated 10 inch filter elements, the choice was made to utilize 47 mm discs, which were flawed by laser drilling to generate a single defined defect of 10 µm diameter.
The 24 g/L trial work was problematic, as the solutions fouled most of the filters rapidly to a degree that the filters could not be wetted as the flow through stopped. However, the data established with 24 g/L foulant all showed that the post-use test was able to detect the flawed filter. In the case of the foulant concentration of 0.8 g/L, two of the fifty-four 47 mm disc filters passed the post-use integrity test at a blockage level of 81% and 97%. Sixteen other filters with a blockage level of >80% failed the post-use test.
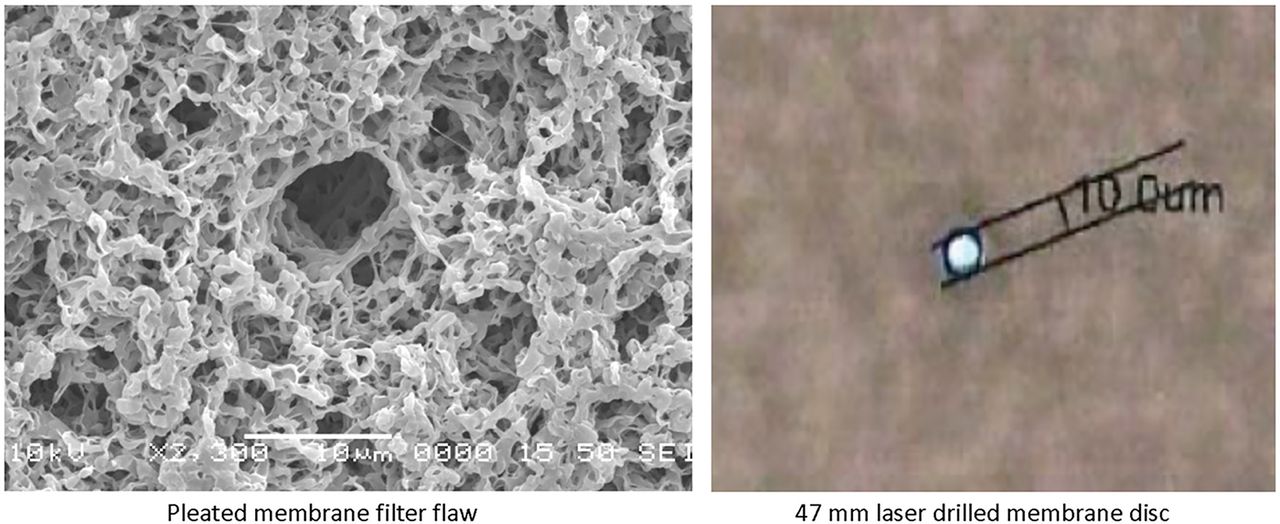
The question may be posed whether the flaws of the pleated 10 inch filter elements and the 47 mm disc flaws were the same, so the test team evaluated the Phase 1 and 2 results and checked the filter flaws microscopically. One of the masked filters was measured as an “abort” bubble point test in Phase 1, which is very much the same result found with the automated integrity tester results of Phase 2. This shows that the flaw in Phase 1 is very similar to the Phase 2 deliberately drilled flaw. In addition, the enclosed microscopic pictures (Figure 1) show that the flaw of the pleated filter device is very similar to the laser-drilled hole in size and shape.

Microscopic images of flawed filter used in Phase 1 and filter with laser-drilled 10 μm holed used in Phase 2.
The test data collected are the first scientific data of the potential of masking sterilizing-grade filters. It was determined that masking is dependent on specific circumstances and can only happen under extreme conditions of either foulant concentration or elevated blockage levels that are much higher than encountered during filter sterilization of drug products.
Recommendation
The data indicate that sterilizing-grade filter masking depends upon product and process circumstances. One requires to understand whether the filtered fluid attributes and process conditions result in a high-fouling filtration process or not. Conditions leading to a high level of filter blockage should be avoided in the final filter sterilization of a drug/medicinal product, and this should be considered during filtration process development and the process validation exercise. The determination of such fouling properties is typically determined by filterability trials. These tests either determine the ideal filter size (effective filtration area) to obtain sufficient filter capacities or pre- and final filter combinations to avoid premature blockage of the final filter. These tests are performed as part of the filter selection program in process development or during the process validation. The activity could also include the determination of the fouling likelihood. The filter fouling potential can be determined by the level of blocking or by the use of deliberately flawed 47 mm disc filters, which would be tested pre- and post-use to see whether the product fouls the filter to the degree of masking.
For terminal sterilizing filtration of many of the biologics, redundant 0.2 µm membrane filtration is used with one of the filters serving as an insurance filter. This means that the filter closest to the filling point is protected by an identical filter and blocking of that filter is highly unlikely to occur.
Ultimately, the need for PUPSIT requires careful determination of the risk of masking a filter flaw, as well as the risk of a flawed filter, which is rare in itself. In the majority of applications, the sterilizing-grade filter is not fouled or blocked, as this would jeopardize the process step, certainly not to the degree of masking. Therefore, a generic enforcement of PUPSIT to all applications of terminal filtration is not scientifically defensible and elevates risk, as the implementation of PUPSIT significantly increases the complexity of the aseptic process and elevates the risk of breaching the sterilized filtrate side of the terminal filter. The points to consider publication describing the best practices of PUPSIT activities points out that PUPSIT is not easy to implement into a process, but requires a multitude of careful determinations (10). In addition, two surveys showed that in almost every case, PUPSIT increased the complexity of the downstream process, of the integrity and filtration process and, with it, elevated the risks.
To compare the universal PUPSIT enforcement, one could think that the entire industry requires to switch to tighter filters, since there have been rare reports of microbial penetration through of 0.2 µm filters. These penetrations happen under specific circumstances and are exceptional. To determine whether this may be the case with the specific product to be filtered at specific process conditions, the end user is required to perform a product-specific bacteria challenge test under process conditions. This process validation step creates assurance that the sterilizing-grade filter is retentive. We have the exact same scenario with PUPSIT, a thorough up-front characterization of the filtration step and process testing along with a risk assessment would be a much better choice than general enforcement.
Conflict of Interest Declaration
The authors declare that they have no competing interests.
Acknowledgments
We would like to express our appreciation to the PDA Training and Research Institute and the laboratory teams of the filter manufacturers for their invaluable support. These efforts would not have materialized without their help. We also would like to thank Wayne Garafola (Sartorius Stedim Biotech), Dr. Rolf Jaenchen (Pall) and Randy Wilkins, Anne Leahy, and Soleil Li (MilliporeSigma) for their advice and test work.
Appendix
Appendix: Study Protocol
Objective
The study protocol describes the test used in the masking studies performed, in which a fluid of two different concentrations that is expected to plug the filter is used to block 10 inch filter cartridges and 47 mm discs from various manufacturers’ 0.2 and 0.45 µm rated membranes made using various polymers and various blocking rates. The masking studies were performed to determine whether a filter with a known flaw that fails a prefiltration integrity test will pass the postfiltration integrity test because the blocking agent is masking the filter flaw.
Test Equipment, Materials & Required Utilities
The following were used to carry out the studies:
Collection of flawed 0.2 µm, 10 inch filter cartridges (Code 7 adapter, 226 double O-Ring, double bayonet end) from all major filter suppliers. The filters were production rejected filters, which failed the integrity test close to the integrity test limits (bubble point and/or diffusive flow).
Collection of 0.2 µm, 47 mm disc filters in which known flaws (e.g., laser-drilled holes) had been deliberately created.
10 inch T-style filter housings with legs, vents, and code 7 adapter (226 O-Ring).
47 mm disc holder.
Pressure transducer with communication to software (typically used for filterability trials).
Scale (50 kg) capacity/flow meter (10 LPM with 0.1 LPM readability) for measuring flow to communicate with software (typically used for filterability trials).
Calibrated integrity test instruments from various manufacturers.
Sartorius Sartocheck 4 Plus.
Millipore IT4.
Pall Palltronic.
Meissner AccuFlux 2.0.
Various pressure vessels for the fluid feed.
0.2 µm filtered deionized water (6 Mohm or better quality).
Filterability kit, which includes all necessary hardware (pressure transducers, filter housing, fittings, gaskets, etc.) required to perform filterability studies.
In-line gas pressure regulator.
Connective tubing, valves, clamps, and collection vessel.
Ovaltine Classic Malt 12 oz.
Access to 120 VAC power.
≥90 psid oil-free compressed air.
Preparation of Standard Masking Formulation
Materials:
Ovaltine powder.
Water: 0.2um filtered deionized water.
Target Concentration: 0.8 g/L and 24g/L.
Mix (minimally, a magnetic mixer/stir bar with a vortex that dimples the liquid surface) for 5 min prior to testing.
The solution is to be used immediately after mixing and cannot be stored.
Test Method
Constant Pressure Testing – 10 inch filter cartridge and 47 mm disc, using an automated filterability test system:
Assemble the test equipment as indicated in the schematic (both the 10 inch filter depicted and a single 47 mm disc filter can be used):
Assemble the data collection system to obtain time, pressure, and flow rate (mass) measurements.
Ensure that all connections are secure and all signal-transmitting components (balance and pressure transducers) are communicating properly with the filterability software.
Install test filter cartridge housing for the 10 inch filter or disc holder using fittings and/or adapters as necessary.
Flush 10 inch filter cartridge or 47 mm disc using water at 20–25°C with the vent valve located on top of the filter housing in an open position. For the 10 inch filter cartridge, purge any air from the upstream side, close the vent valve, and flush system with water until 50 L/m2 (35 L/0.7 m2) has been processed by the test filter cartridge or longer to meet the manufacturer’s minimum wetting requirements. To provide additional assurance that the wetting fluid fully wets the entire membrane structure, the downstream side of the filter housing may be flow restricted to 3–5 psi to assure that the wetting fluid reached the entire membrane structure. For the 47 mm disc, purge any air from the upstream side, close the vent valve, and flush the system with water according to the manufacturer’s minimum specifications.
Perform pre-use WATER wet integrity tests of the filter, using one of the different filter manufacturer’s integrity test instruments that tests and reports both the diffusive flow and the bubble point test value using compressed air. Record the quantity of the liquid used to flush the filter prior to testing, the measured pre-use integrity test values, the temperature of the wetting fluid, the temperature of the surrounding air, and the integrity test instrument used. Upon completion of the integrity test, drain any excess liquid from the system by opening valves V-02, V-03, and V-04 from the schematic and allow any residual liquid present to drain (do this without removing the filter element from the housing).
Select and prepare one of the two standard masking formulations (0.8 g/L or 24 g/L Ovaltine) to be used in the test just prior to the testing for each filter.
Fill the pressure vessel with one of the standard masking formulations and set the air regulator supplied to the pressure vessel to maintain 10 psig of top pressure on the vessel. Prime the test filter system by introducing product with the vent valve located on top of the filter housing in an open position. Purge any air from the upstream side, and close the vent valve once all the air has been evacuated. This indicates that the test system is devoid of any air bubbles that would prevent access to the filter membrane. Refill the vessel with fresh standard masking formulation after each masking test performed.
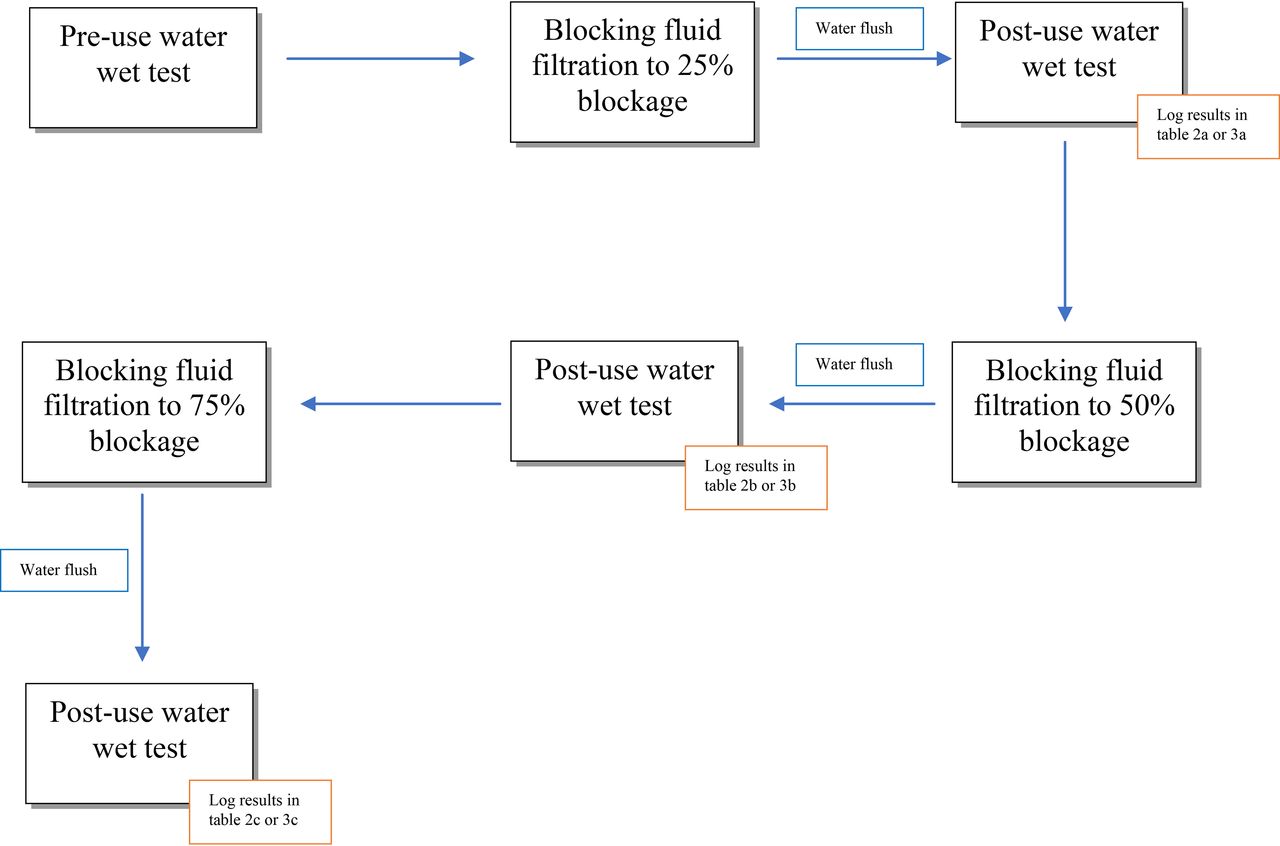
Run each filter trial using the standard masking formulations until the flow rate decreases to 75% of the initial flow rate (25% flow decay). Record the total processed volume of the standard masking formulations.
Flush the standard masking formulation from the filter using a water. Flush the filter cartridge or disc using water at 20°C–25°C with the vent valve located on top of the filter housing in an open position. Purge any air from the upstream side, close the vent valve, and flush the system with water until 50 L/m2 (35 L/0.7 m2) has been flushed through the filter.
Perform post-use WATER wetted integrity tests of the filter. Using the same integrity test instrument used previously on this filter, perform an automated integrity test that reports both the diffusive flow and bubble point test values with compressed air. Note: ensure that the correct test parameters are selected for the integrity test being performed (e.g., the pressure that the diffusion flow rate is determined at, the acceptance criteria for diffusion and bubble point for the filter tested). Record the quantity of water filtered through the filter to flush out the standard masking formulations prior to testing, the measured post-use water wetted integrity test values, the temperature of the wetting fluid, the temperature of the surrounding air, and the integrity test instrument used.
Run each filter trial again using the standard masking formulation until the flow rate decreases to 50% of the initial flow rate (50% flow decay). Record the total processed volume of the standard masking formulation.
Flush the standard masking formulation from the filter using water. Flush the filter cartridge or disc using water at 20°C–25°C with the vent valve located on top of the filter housing in an open position. Purge any air from the upstream side, close the vent valve, and flush the system with water until 50 L/m2 (35 L/0.7 m2) has been flushed through the filter.
Perform post-use WATER wetted integrity tests of the filter. Using the same integrity test instrument used previously on this filter, perform an automated integrity test that reports both the diffusive flow and bubble point test values with compressed air. Note: ensure that the correct test parameters are selected for the integrity test being performed (e.g., the pressure that the diffusion flow rate is determined at, the acceptance criteria for diffusion and bubble point for the filter tested). Record the quantity of water filtered through the filter to flush out the standard masking formulation prior to integrity testing, the measured post-use water wetted integrity test values, the temperature of the wetting fluid, the temperature of the surrounding air, and the integrity test instrument used.
Run each filter trial again using the standard masking formulation until the flow rate decreases to 25% of the initial flow rate (75% flow decay). Record total processed volume of the standard masking formulation.
Flush the test formulation from the filter using water. Flush the filter cartridge or disc using water at 20°C–25°C with the vent valve located on top of the filter housing in an open position. Purge any air from the upstream side, close the vent valve, and flush system with water until 50 L/m2 (35 L/0.7 m2) has been flushed through the filter.
Perform post-use WATER wetted integrity tests of the filter. Using the same integrity test instrument used previously on this filter, perform an automated integrity test that reports both the diffusive flow and bubble point test values with compressed air. Note: ensure that the correct test parameters are selected for the integrity test being performed (e.g., the pressure that the diffusion flow rate is determined at, the acceptance criteria for diffusion and bubble point for the filter tested). Record the quantity of water filtered through the filter to flush out the standard masking formulation prior to testing, the measured post-use water wetted integrity test values, the temperature of the wetting fluid, the temperature of the surrounding air, and the integrity test instrument used.
Perform additional filter trials as previously using the standard masking formulation to further decrease the flow rate below 25% of the initial flow rate (75% flow decay).
Repeat steps 1–18 for each filter and the two different standard masking formulations.

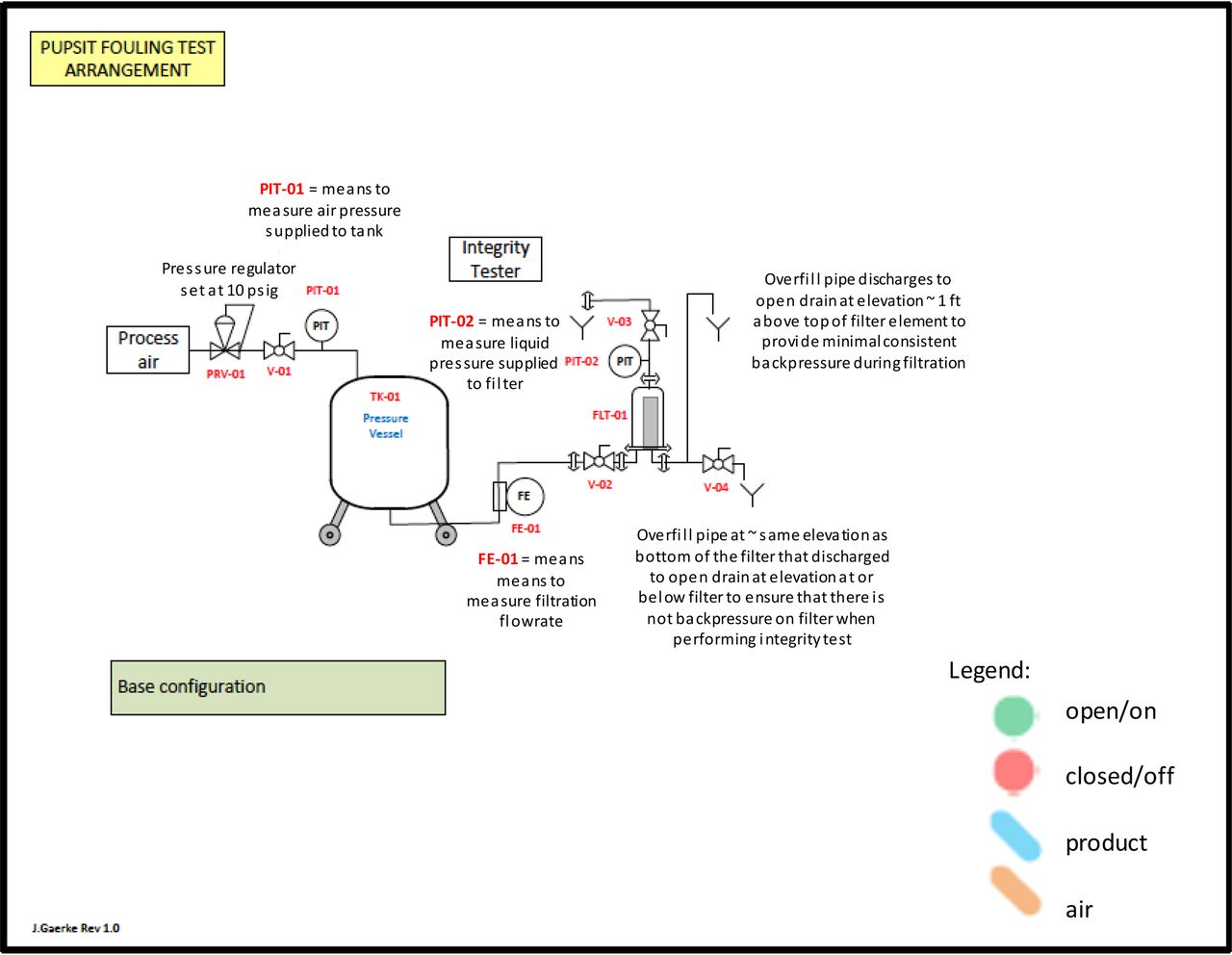
Masking trial test assembly.

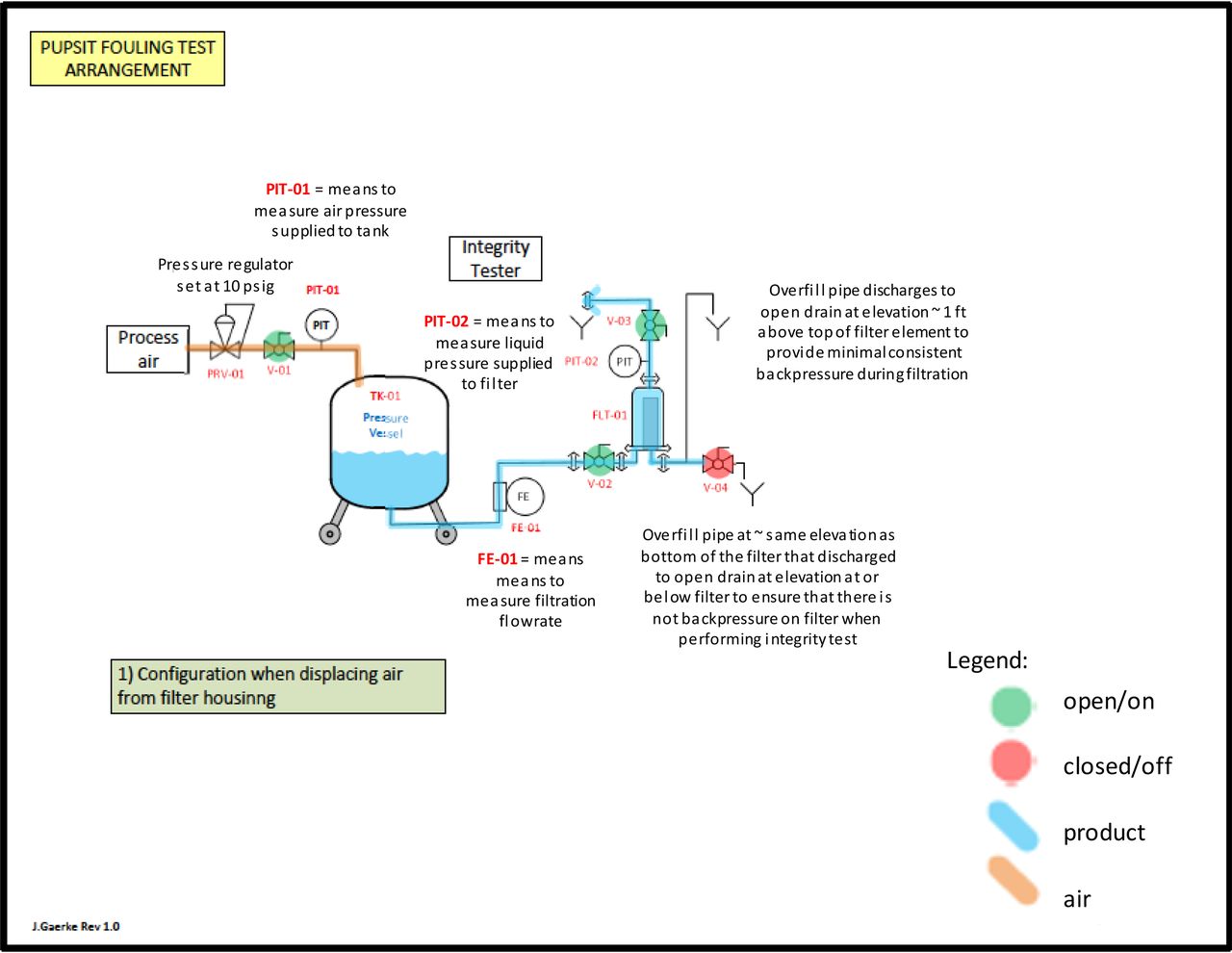
Masking trial test system when venting air from the system.
Schematic of Testing:

Masking trial test assembly during the filtration process.

Masking trial Phase 2 blocking and integrity test schematic.
- © PDA, Inc. 2020




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}