
Abstract
The article proposes an implementation road map of a contamination control strategy (CCS) in a facility. The CCS is the culmination of an exercise to identify activities designed to prevent microorganism, pyrogen, and particulate contamination in the product, the facility, and the supporting processes used to manufacture the product. Manufacturers can formulate their contamination control strategy based on information in the quality target product profile or in the critical quality attributes, in the facility, and in the processes used to manufacture and transport the product. The strategy implementation involves executing the strategic plan and managing the implementation by priority overtime should it be deployed. The evaluation of the efficiency and effectiveness of the contamination control strategy implemented is confirmed by analyzing and trending the various quality performance parameters related to contamination control. The strategy evaluation allows the manufacturer to identify a new strategic plan to support improvement goals or new measures and/or controls to achieve the desired result, minimizing the contamination risk.
Introduction
The term contamination control strategy (CCS) was introduced in the recent changes of the European Union (EU) draft Annex 1 version 12 (1). Draft Annex 1 proposes a similar definition of CCS to the control strategy definition in the International Conference on Harmonisation (ICH) Q10 (2). However, the set of controls should focus on contaminations related to particles, pyrogens, and microorganisms derived from current process understanding that ensures process performance and product quality.
The principle of contamination control or prevention of microbiological, particulate, or pyrogen contamination is not new and is significantly discussed in the regulatory and industrial guidelines (3⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓–25). Therefore, likely most manufacturers have documents that discuss contamination control program per process. However, they may not have a holistic or a single document summarizing all the critical control points to assess the effectiveness of all the controls and monitoring measures employed to manage risks associated with contamination across a facility or in the final product.
The CCS main goals are to (1)
identify the set of controls required to detect and prevent microbial, pyrogen, and particulate contamination across the facility and in the final product
assess the collective effectiveness of all the controls and monitoring measures employed to prevent the risk of contamination across the facility (e.g., utilities, cleaning and disinfection, process validation, facility design, etc.) and in the final product
improve the quality system with continuous improvement plans based on the analysis and trending of data gathered through the monitoring measures employed
assess the evolution of the contamination control performance over time
Consequently, the CCS development and its documentation require robust technical, process, and contamination control expertise (1). This article proposes an implementation road map of a CCS across a facility. It adds to existing publications (26⇓⇓–29) by proposing a road map to design a holistic and transversal document that identifies all the controls (design, procedural, technical, and organizational) and monitoring measures employed to manage risks associated with contamination, to evaluate the performance, and to continuously improve the CCS.
Implementation Road Map
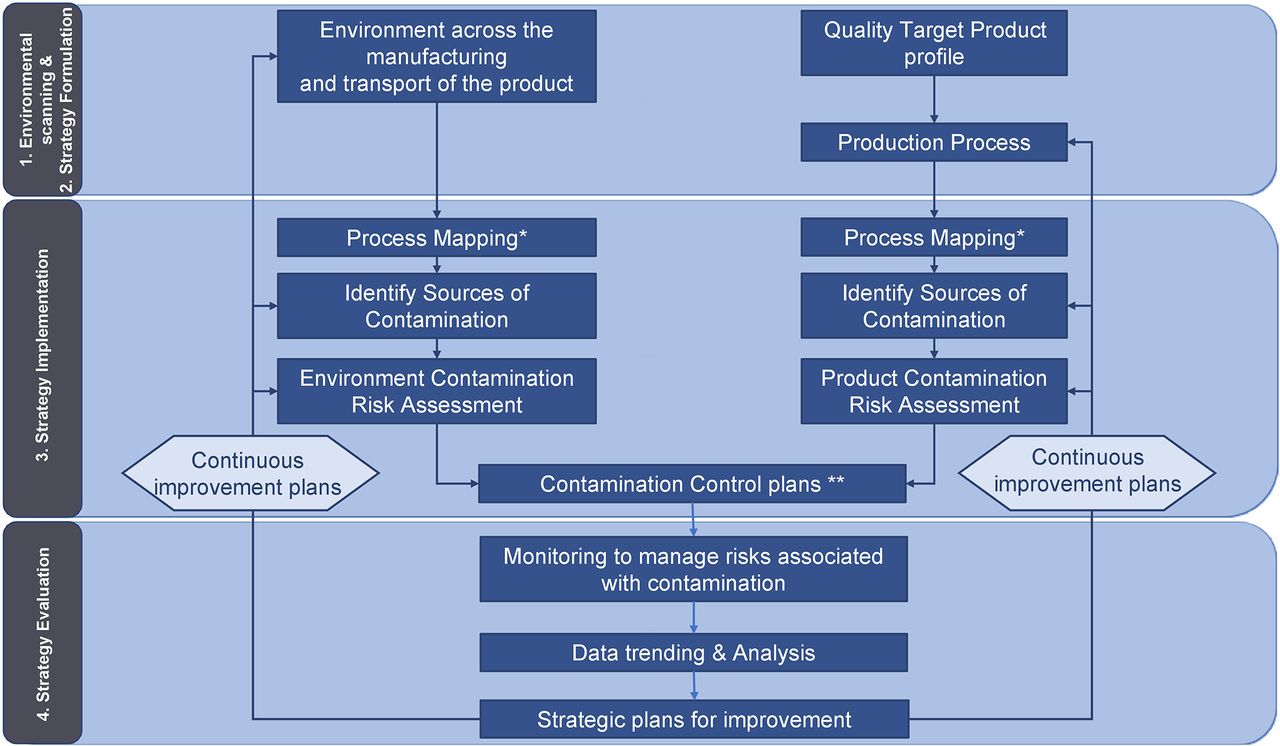
The implementation of a CCS consists of four steps (Figure 1).
Environmental scanning: In this article, “environmental” or “environment” refers to all the design, procedural, technical, and organizational elements needed to manufacture the product (e.g., facility design, cleaning and disinfection, utilities, equipment, sterilization, depyrogenation, aseptic manipulation, etc.). “Scanning” refers to a process of collecting, scrutinizing, and providing information to formulate the strategy.
Strategy formulation is the process of deciding the best course of action for accomplishing the desired results.
Strategy implementation implies making the strategy work as intended by implementing the organizational activities, procedures, controls, monitoring, resources, and decision-making tools, and more.
Strategy evaluation measures the performance of the processes and confirms the strategy put in place to achieve the desired results.

Contamination Control Strategy: Implementation Road Map. *For a new process, the mapping is replaced by process designing. **including design, procedural, technical, and organizational implementations.
The content of the CCS could be documented following the four steps listed earlier.
Environmental Scanning
Thorough scanning of the environment where the product is produced and transported is essential to identify the level of effort and formality of the CCS implementation and documentation. The scanning will also support the CCS formulation based on the type of product (sterile or nonsterile) or intermediate (sterile, bioburden control, or nonsterile) produced in the facility.
The scanning should first analyze the type of products or intermediate (for example, active pharmaceutical ingredient, drug substance, intermediate product) manufactured in the facility. When a drug product is manufactured, the Quality Target Product Profile (QTPP) of the finished product should be analyzed (30). The QTPP contains a summary of the quality characteristics of a drug product. Some of the quality characteristics (for example, drug product quality criteria and microbial attributes) may be useful to formulate and assess the complexity and the formality of the CCS implementation. The quality characteristics contain information on the product release specifications such as sterility, bioburden, endotoxin, and particulate limits. Also, critical quality attributes (CQA) of a product or an intermediate are generally derived from the QTPP information (30). Consequently, for an intermediate manufactured in a facility, the scanning step should examine the CQA (30).
The scanning should also collect and scrutinize all the elements to be considered within the CCS documentation, such as, but not limited to, the design of the plant, type of product manufactured in the plant, premises, equipment, personnel, utilities, cleaning and disinfection, sterilization, and so forth (1).
The complexity and formality of the CCS implemented in a facility may differ depending on the microbial, endotoxin, and particulate specifications of the product or intermediate manufactured.
The implementation of the CCS for a nonsterile drug product is not mandatory (1). However, suppose a nonsterile drug product manufacturer elects to apply some principle and guidance on contamination control strategy. In that case, the manufacturer should document which principle has been applied and confirm compliance with that principle (1).
Contamination Control Strategy Formulation
Several elements must be understood and analyzed to formulate a robust and effective CCS (1, 2, 26, 27). The EU Good Manufacturing Process (GMP) draft annex 1 version 12 paragraph 2.5 suggests several elements (not an exhaustive list) to consider when developing the CCS (1).
The CCS document's strategy formulation section should present the strategy, the facility vision, and the strategic plan. The strategic plan contains a list of actions to be implemented by priority and deadline. The document details the steps needed to reach the desired results of each identified action. The implementation's priority may depend on its impact on the CCS performance and the cost to implement. Some actions may require significant capital expenditures.
Contamination Control Strategy Implementation
The strategy implementation consists of executing the strategic plan. The strategic plan's execution may require the implementation of control strategies, procedures, processes, and organizational elements to prevent contamination. The mapping of the pharmaceutical processes helps identify contamination sources and assess the residual risk of contamination. A gap analysis comparing the residual risk against the facility CCS objective will help define the control strategies, procedures, processes, and organizational elements to implement.
The mapping of pharmaceutical processes is critical to defining the elements of the risk assessment and to developing process knowledge (27, 29, 31, 32). By identifying the sources of contamination, the manufacturer can evaluate the risk of contamination in the processes. It is acceptable to use existing quality risk assessments (QRAs) such as microbial, bioburden, particulate, or product quality risk assessment. However, the manufacturer must ensure that the documents cover the CCS's purpose and are still up to date. The CCS's goal is to identify controls triggered by microorganism, pyrogen, and particulate contamination in the product or the surrounding environment; therefore, the pharmaceutical manufacturer should ensure that their existing QRA covers those elements.
QRA must be used to define the element of control strategy (Figure 1). The controls can include parameters and attributes related to drug substance, excipient, drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control (1). The control strategy can also include parameters such as vendor qualification, preventive maintenance program, and Corrective Action Preventive Action (CAPA) effectiveness to improve the CCS performance.
The monitoring location and frequency are implemented according to the control strategy and the risk of contamination (1, 26). The historical monitoring data must be collected, analyzed, and trended to confirm the process performance and product quality over time (33). The trends analysis should also include data from root cause investigations due to deviations.
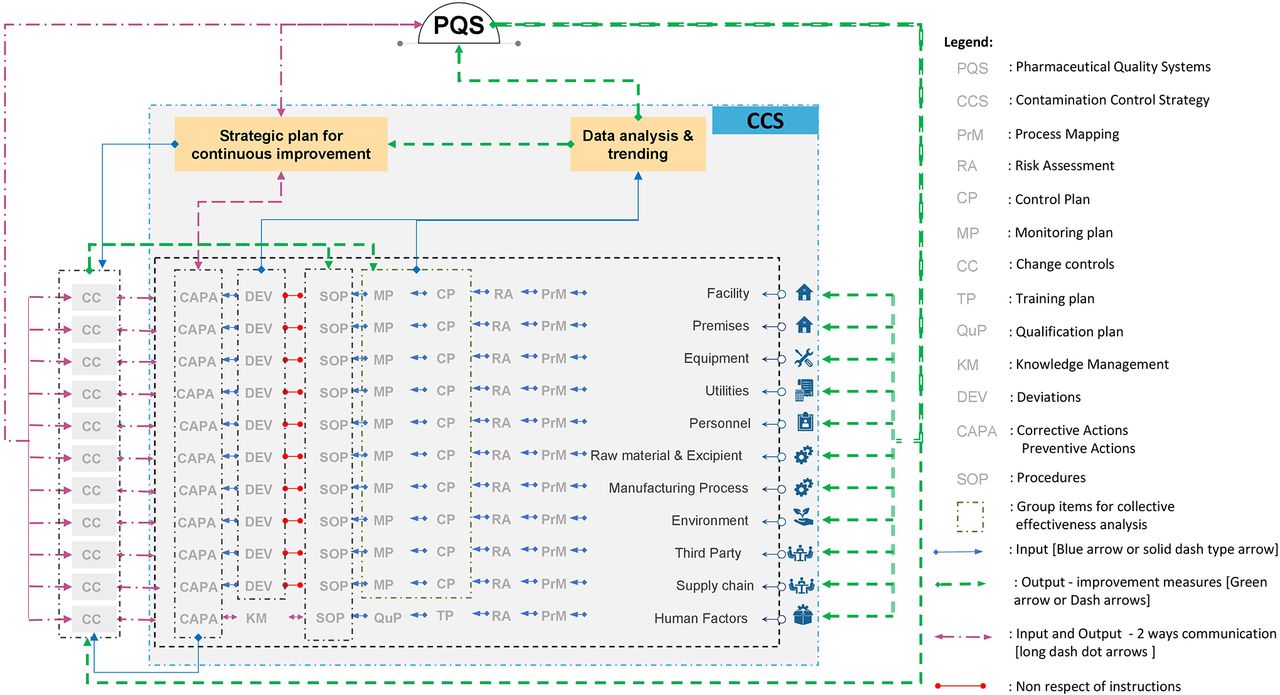
The development of the CCS requires thorough technical and process knowledge to be developed and implemented across the facility to define all critical control points and assess the effectiveness of all the controls (design, procedural, technical, and organizational) and monitoring measures employed to manage risks associated with contamination (1). The CCS should consider all aspects of contamination control with an ongoing and periodic review (data analysis and trending; Figure 2), resulting in updates (strategic plan for continuous improvement; Figure 2) within the quality system as appropriate (1). The collective effect of the measures and controls taken to minimize the risk of contamination should be considered altogether.

CCS implementation across a facility. The model starts with the mapping process (PrM) of the different elements to manufacture and transport the product. The mapping of the processes will ultimately help in identifying the sets of control (CP) and the inputs (blue arrows and pink arrows when in direction to the PQS or “strategic plan for improvement” box) needed to feed the design and evaluation of the CCS. The analysis and the trending of the input data will lead to output (strategic plan) to improve the PQS, the processes in place to prevent contaminations (green arrows or pink arrows when going back to the different elements such as facilities, premises, utilities, etc.). Note that third party refers to, for example, outsourced activities, suppliers, etc. Personnel refers to personnel flows, intervention, activities, etc. Human factors refer to behavior, hygiene, training, qualification, disqualification, etc.
Communication of the CCS strategy, implementation, and updates to fill gaps or to drive continuous improvement may be performed by a cross-functional team. This cross-functional team may be composed of a QRM facilitator, quality assurance, a senior microbiologist expert, and a process expert supported by other departments (29, 34). The communication includes feedback from the manufacturing staff when gaps or improvements are identified. Decision makers (for example, the senior management team) should also communicate their decisions to ensure prioritization of the measures deployed and maintenance of the CCS level (35).
The CCS implementation section may contain or cross-reference existing documents (for example, quality risk assessment, design specifications, control strategy) that list and provide a rationale for
all the controls, including
design (i.e., facility design; heating, ventilation, and air conditioning (HVAC) control; equipment design)
procedures (i.e., cleaning and disinfection, sterilization, aseptic manipulation)
technical (i.e., preventive maintenance, technologies implementation)
organization (i.e., resource allocations, work shift, decision-making process, training)
samples or monitoring measures employed to be trended and analyzed
communication and implementation process of the CCS
team composition
Contamination Control Strategy Evaluation and Improvement
The strategy evaluation uses the historical performance and data trend analysis to shed light on the CSS's efficiency and effectiveness in achieving the desired results. The strategy evaluation must consider the evolution of the overall risk priority number (RPN) value (36) of the residual risk of each process over time (t) or over several batches produced (for example, campaign) (Table I). The goal is to demonstrate the effectiveness of the improvement implemented (strategic plan) from the previous evaluation or trigger immediate actions to prevent product contamination. Therefore, pharmaceutical manufacturers should define the RPN value of the residual risk that would trigger an immediate action.
Strategic Control: Example of a Control Dashboard to Confirm the CCS Effectiveness over Time (t)
Note that other quality performance parameters (QPP) triggered by microorganisms, pyrogens, or particulate contamination could be used to track the CCS performance (37), for example,
% of deviations
% of rejected batches or % of write-off
% of negative environmental monitoring trends
% of recalls
% of sterility or microbial (total aerobic microbial count, total combined yeasts and molds count for nonsterile) final release testing failure
% of complaints received
% of exceeding limits in grade A (ISO5)
% of failed aseptic media simulation
% of disqualified aseptic operators
% of actions outlined in the strategic plan that has been implemented during a defined period.
A collective QPP analysis at a regular frequency should be considered to evaluate and improve the CCS performance. The evaluation frequency depends on the overall contamination risk level, product quality risk, and any changes that may impact the CCS level.
The CCS evaluation and improvement section may contain the RPN value of the residual risk and the QPPs that are evaluated at a defined frequency. It should also include the improvement that needs to be implemented based on the data analysis.
Conclusion
Manufacturers can formulate their contamination control strategy based on the quality target product profile or critical quality attributes, the facility, and the processes used to manufacture and transport the product. The strategy implementation involves executing the strategic plan and managing the implementation by priority over time. The strategy evaluation uses the historical performance and data trend analysis to shed light on the efficiency and effectiveness of the contamination control strategy. The strategy evaluation allows the manufacturer to identify a new strategic plan to support improvement goals or new measures and controls to achieve the desired result, minimizing the contamination risk.
Conflict of Interest Declaration
The author declares that he has no competing interests.
Acknowledgments
The author is grateful for his colleagues, who provided several valuable contributions that significantly improved this article.
- © PDA, Inc. 2021




{kind=link}
{kind=link}