
Abstract
The number of products being developed for serious conditions that are eligible for expedited programs has been increasing in recent years. This article presents an industry perspective on how to reduce analytical life cycle steps when using analytical platform technologies (APT) in support of accelerated biological product development. Strategies for life cycle steps for APT methods are conceptually reviewed within the framework of supporting chemistry, manufacturing, and control (CMC) development acceleration. Reduced method qualification, transfer, and validation studies could be performed, provided that the initially validated test method remains unchanged. A detailed case study is used to illustrate considerations for the initial method validation and subsequent APT verification studies. Considerations for APT implementation are discussed and suggestions are provided for the submission of APT information in regulatory filings.
1. Introduction and Scope
Industry and regulators are looking for ways to accelerate product development as the number of products being developed for serious conditions that are eligible for expedited programs has been increasing (1). Following the California Separation Science Society (CaSSS) chemistry, manufacturing, and control (CMC) Strategy Forum presentations in 2014 (2⇓–4), the author was invited to present an industry perspective on how to reduce analytical CMC steps in support of accelerated biological product development to the Center for Drug Evaluation and Research (CDER)/U.S. Food and Drug Administration (FDA) in 2015 (5–6). This presentation also followed up on a 2012 presentation to CDER by the author, which covered the content from Parenteral Drug Association Inc. (PDA) Technical Report No. 57: Analytical Method Validation and Transfer for Biotechnology Products (7–8).
In this article, strategies for life cycle steps for analytical platform technology (APT) methods are conceptually reviewed within the framework of supporting CMC development acceleration. An APT method is an analytical method used for multiple highly similar products or product sample matrices without modification of the procedure. Suitable for use as APT methods are those methods that by design and intent can generate accurate and reliable results without any significant product-specific interference. For example, separation test methods for monitoring product impurities and product variants are typically suitable for APT use whenever sufficient peak separation and the lack of potential matrix interference has been established over a wide range of potential impurity results expected. Similar to compendial methods, an approved APT method may not require full validation for each new product or sample type. A test method becomes approved as a platform method once the relevant supporting data has been reviewed and authorized as part of a marketing application.
A detailed case study is used to illustrate considerations for the initial development and validation studies of an APT method. The submission details as well as other relevant practical considerations are based on the meeting outcome with CDER (5). Although suggestions are provided for how to submit APT information in regulatory filing, each regulatory jurisdiction may have specific requirements.
In this article, the concept and practical considerations for implementing APTs are built upon the International Conference for Harmonisation (ICH) Q2(R1) guidelines (9) and PDA Technical Report No. 57 (8). Similar to manufacturing process validation, analytical method validation (AMV) can then be defined as the collection and evaluation of data, from the analytical method development (AMD) throughout routine quality control (QC) testing, which establishes scientific evidence that an analytical method is capable of consistently delivering accurate and reliable results (8).
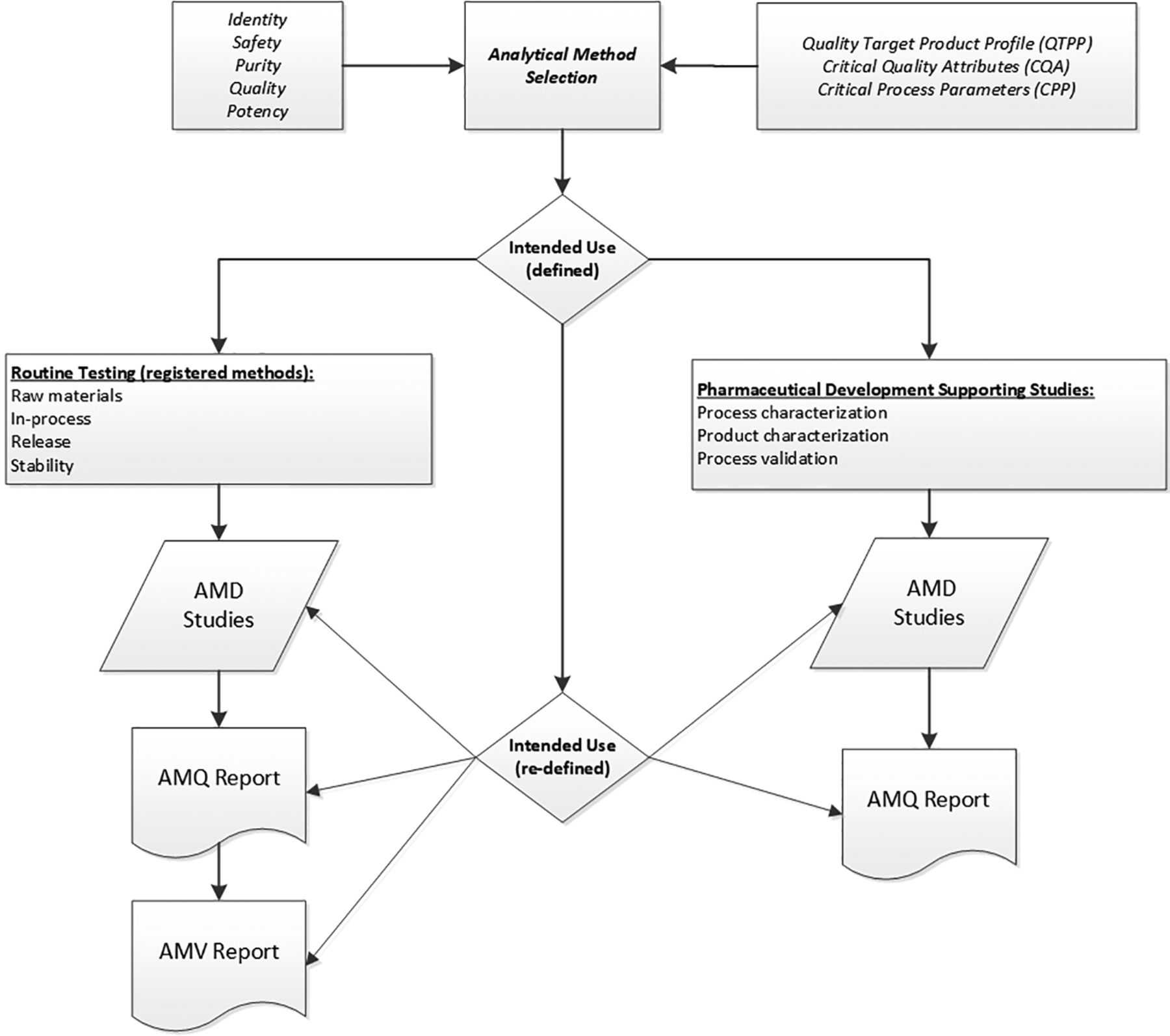
AMV studies are typically executed for product quality specification test methods but may not be required for analytical methods used in support of pharmaceutical development. Figure 1 illustrates the life cycle path of the two different analytical methods. They are separated according to the intended use of a particular method (8), which is assessed early in development as part of the overall quality target product profile (QTPP).

Method life cycle from initial QTPP through post-AMV studies. AMD, analytical method development; AMQ, analytical method qualification; AMV, analytical method validation; QTPP, quality target product profile.
2. Risk-Based Validation Concepts
The rigor of prospective AMV studies can be assessed by differentiating these studies into four general AMV categories (A–D). The four categories or types of studies come into effect depending on each of four possible general situations as summarized in Table I (8). Risk ratings are only qualitative and are primarily based on the level of product quality variation and the confidence in the historical test method performance. In general, the less is known about future product variation and/or test method performance, the greater the associated level of uncertainty and resulting risk levels.
AMV Categories and Prospective Validation/Verification Studies (7)
AMV categories are listed in order of decreasing risk and/or uncertainty level(s). Category A typically has the greatest uncertainty and risk to patient and/or firm because the relationship between the product/process and the analytical method performance capabilities may not be fully understood. Typically, the greater the risk, the more data needs to be generated to offset it. Previously validated and approved APTs (AMV category C) would typically require fewer prospective AMV studies, because the historical method performance is well captured. In the case of Category D (compendial methods), minimum prospective (verification) studies are required, because their use and suitability has been extensively demonstrated. However, prospective verification studies under actual conditions of use with representative test samples are still required for compendial methods.
Table I is only a general summary of the existing AMV categories and typically associated qualitative risk levels. In practice, the actual risk/uncertainly levels for specific test methods may significantly vary for particular test methods. For example, the risk level for the verification (or validation) for category D of the compendial Sterility and/or Bacterial Endotoxin Test (BET) may be higher than indicated in Table I. The severity of the potential impact on the patients for false-negative test results is highly significant for safety tests. An example for higher risk is an unexpected low endotoxin recovery (LER) study result as part of the overall BET method suitability studies.
3. Accelerated Product Development
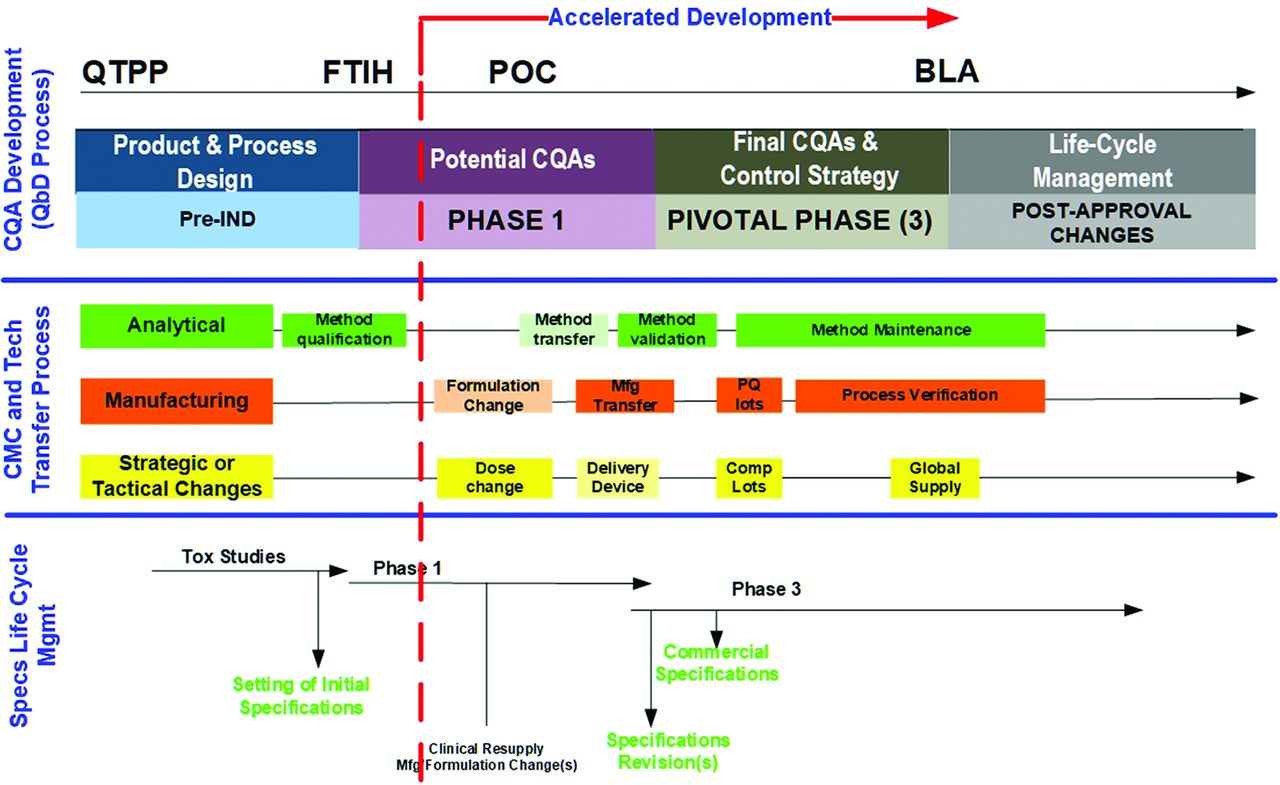
The decision by the regulators to permit the acceleration of a development program, whether through an accelerated approval, a fast track, or a breakthrough product designation, will significantly reduce the product and method development time line. Using prior/platform knowledge is essential for successful, rapidly moving CMC and clinical programs. An accelerated development program for a new investigational medicinal product (IMP) is illustrated in Figure 2. In this example, a product development program is accelerated after the results from the early-stage clinical studies showed results consistent with promising preclinical study results. The significant reduction of clinical studies and the associated duration of these trials leads to the acceleration of the typical CMC development steps to remain aligned with the now much closer license submission target date. In this accelerated scenario, essentially all typical parallel or sequential CMC steps, including all AMV process steps, require much faster completion.

Accelerated CQA development, CMC changes, and specifications. BLA, biologics license application; CMC, chemistry, manufacturing, and control; CQA, critical quality attribute; FTIH, first time in human; IND, investigational new drug; POC, proof of concept; PQ, process qualification; QbD, quality by design; QTPP, quality target product profile.
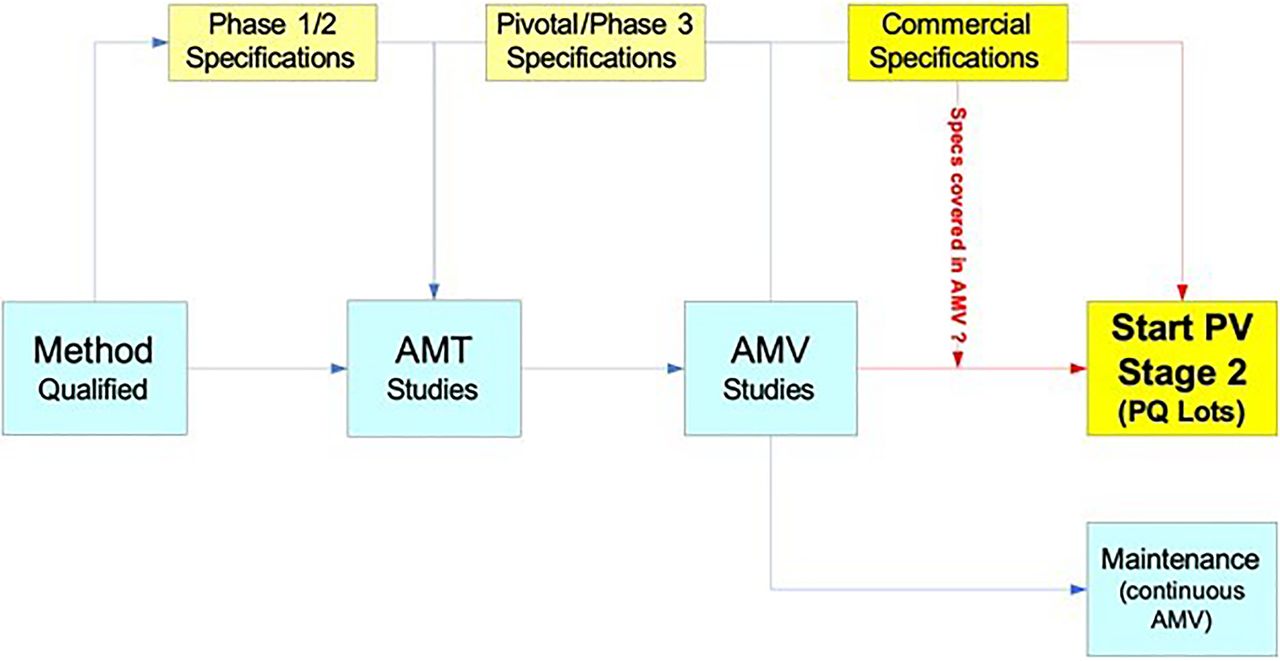
The typical analytical method life cycle steps within the phase-appropriate specification revision cycles is illustrated in Figure 3. The AMV process can be considered, similar to manufacturing process validation, to consist of three validation stages (8). The first stage is when the method development, qualification, optimization, and robustness studies typically occur. Similar to manufacturing process validation, stage 2 is when formal AMV studies are completed, and stage 3 is the test system performance monitoring and control program (“method maintenance”). Formal AMV studies (AMV stage 2) are completed before the manufacturing of the process qualification (PQ) lots to support the process validation (PV) stage 2 campaign and specifications. During and following PV stage 2, the continuous AMV program (AMV stage 3) starts.

Typical analytical method and specification life cycle(s).
Note: Analytical method transfer (AMT) can occur at any product development stage. In this illustration, AMT occurs prior to pivotal phase manufacturing transfer and analytical method validation (AMV) studies. AMT, analytical method transfer; AMV, analytical method validation; PQ, process qualification; PV, process validation.
Regulatory expectations for the completion of an AMV study may vary depending on the product type and quality attribute tested. Typically, AMV studies are executed against the current product quality specifications established before PV. This is usually initiated during the pivotal clinical phase studies. AMV studies are usually completed 1-2 years before application for a commercial license for a standard CMC development program. Depending on the speed and acceleration of the product development and how much real-time stability data is desired following the PQ runs (PV stage 2), the AMV studies may be completed much closer to license submission.
It should be noted that AMV studies are often completed before the PQ specifications are internally approved. The quality system procedures, covering the specification life cycle, should contain a “check-back” requirement whether the AMV studies are still valid for the final specifications (8). Typically, quantitative specifications are tightened for later-stage clinical and commercial drug substance and drug product. In those cases, the completed analytical method validation studies will have covered at minimum the specification range(s). However, in cases in which additional specification limits are set, analytical method qualification (AMQ) or AMV studies may require additional studies to cover this major specification revision.
4. APT Concept and Use Conditions
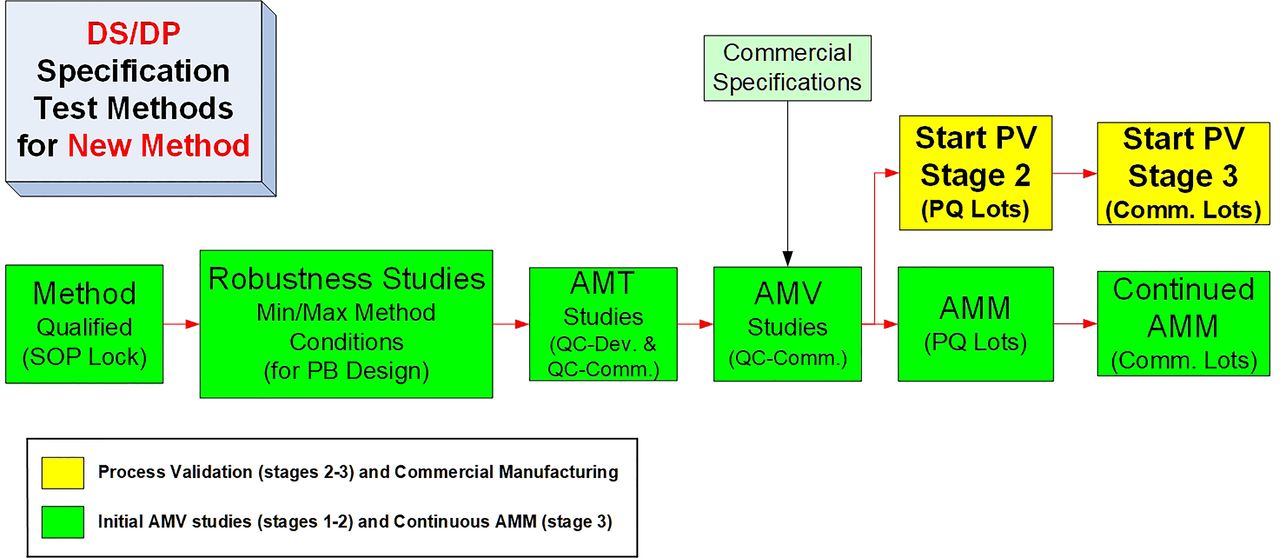
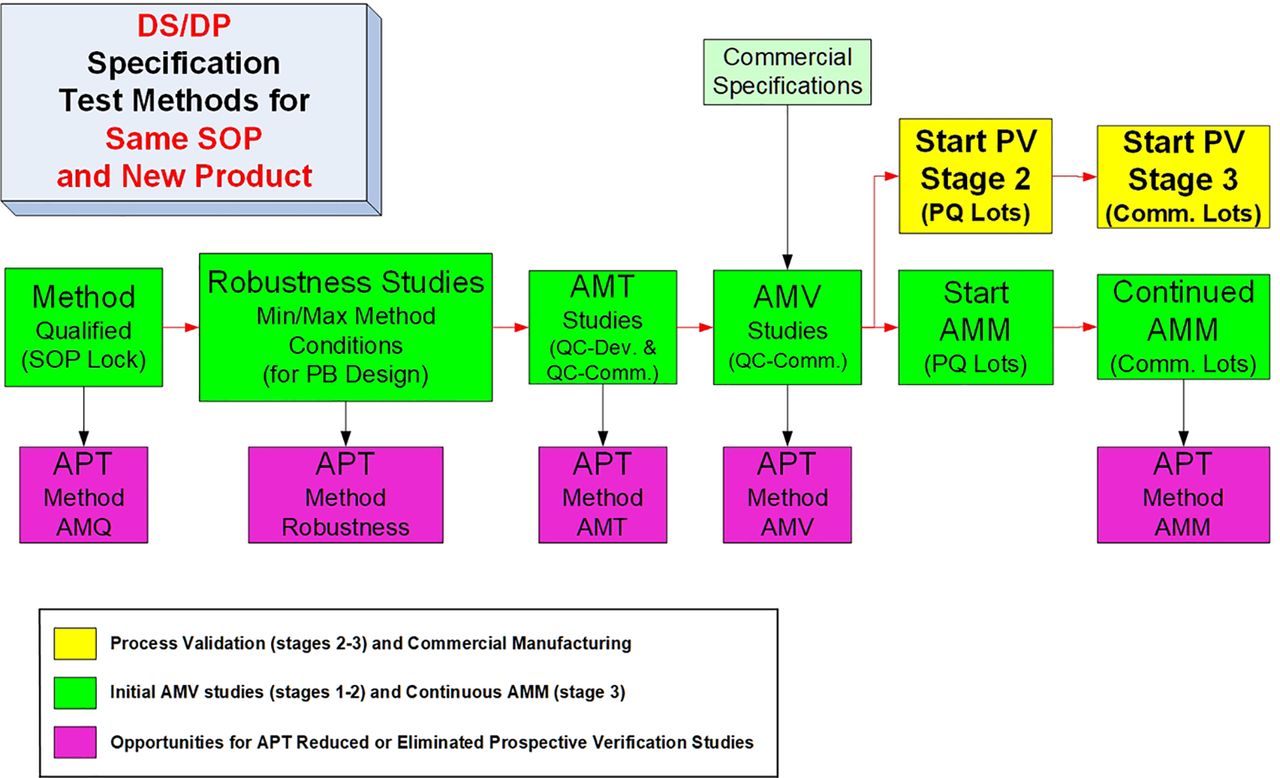
A more detailed and ideal sequence for the life cycle of a new test method is shown in Figure 4. The illustrated AMV process can be considered to be an ideal step sequence, because the test method standard operating procedure (SOP) is locked with the AMQ step, and all release/stability data for investigational medicinal product (IMP) is generated by a qualified method, confirmed for suitability of its intended use. The method robustness studies are executed after the completion of any necessary method optimization steps but before the analytical method transfer (AMT)/AMV study execution so that an optimized, robust test method is then validated. The AMV studies are then executed before the PQ studies to ensure the PQ studies are performed with validated test methods. This will then mostly remove the remaining analytical uncertainty when conducting the PQ studies. Similar to the CPV stage (PV stage 3), the analytical method maintenance (AMM) program can then start with the first PQ lot tested.

Typical life cycle steps for a new routine test method.
AMM, analytical method maintenance; AMT, analytical method transfer; AMV, analytical method validation; DS, drug substance; DP, drug product; PB, Plackett-Burman; PQ, process qualification; PV, process validation; QC, quality control; SOP, standard operating procedure.
The additional detail provided can then be used to evaluate and illustrate which life cycle steps can be reduced for APT methods. In Figure 4, the method is qualified before IMP release testing, and the SOP is then “locked” until changes become necessary. For specific guidance regarding AMQ studies and post-AMQ steps, see PDA TR 57-2 (10) and PDA TR 57 (8), respectively. Method robustness studies can be completed any time post-AMQ and pre-AMV. However, completing robustness studies early will result in less overall risk for completing rapid and successful AMT or AMV studies. Avoiding min/max conditions and/or other contributing variation factors observed in robustness studies may lower the confidence of historical test results.
For the initial AMV studies, the method life cycle steps are typically executed sequentially. As current drug substance/product (DS/DP) specifications frame the intended use of the test method and thus drive the AMV study design and acceptance criteria, it is therefore a critical confirmation step that the suitability of the method’s intended use can be extended into any post-AMV DS/DP specification revisions (8).
Following the successful AMV study completion, the AMM (AMV stage 3) is then typically started with test results from the PV stage 2 studies. The method validation stage 3 is practically initiated before continuous process verification (CPV), which is formaly initiated with the first postlicensure manufactured lot. Upon receiving marketing authorization (e.g., Biologics License Application [BLA]), the “approved” method can now become an APT method. If used unchanged for a similar new development product, each of the five life cycle steps can be significantly reduced or eliminated as illustrated in Figures 5 and 6.

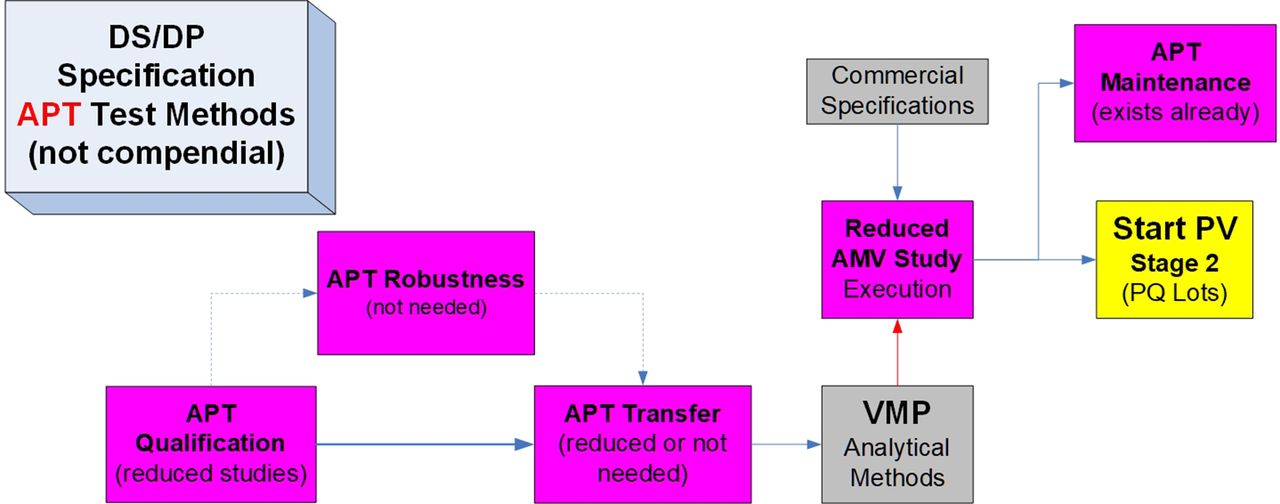
APT opportunities following AMV study completion and BLA approval.
AMM, analytical method maintenance; AMQ, analytical method qualification; AMT, analytical method transfer; AMV, analytical method validation; APT, analytical platform technology; BLA, biologics license application; DS, drug substance; DP, drug product; PB, Plackett-Burman; PQ, process qualification; PV, process validation; QC, quality control; SOP, standard operating procedure.

Risk/uncertainty levels and risk-based opportunities (typical).
AMV, analytical method validation; APT, analytical platform technology; DS, drug substance; DP, drug product; PQ, process qualification; PV, process validation; VMP, validation master plan.
Starting with the AMQ studies for the approved APT method, all possible reduction opportunities for prospective studies for all five method life cycle steps are summarized. As some or most of the initially completed prospective study results are used as the foundation for each of the validation stages, the additional prospective verification studies are proposed here:
(Prospective Study) APT Reduction Opportunities for Method Life Cycle Steps(s):
AMQ—Similar to AMV studies (see following), AMQ studies can be reduced to confirm that different protein concentration(s) or formulation(s) do not impact significantly the validated characteristics accuracy (and inferred specificity). A spiking study below/above DS/DP specification(s), detection limit (DL), or quantitation limit (QL), as relevant, can be used.
Robustness—Previous robustness results can be used without repeating this study, because the method has remained unchanged.
AMT—Previous AMT results can be used, because the method has remained unchanged and has been in routine use at the receiving laboratory since the initial transfer.
AMV—See AMQ prior and also in the case study following.
AMM—Stage 3 of AMV was already initiated at the receiving laboratory for this method. All relevant test system controls have already been established.
Looking for additional opportunities to support a highly accelerated product development process, several options exist. For example, the AMT and AMV studies could be combined to reduce the time to complete AMV stage 2. Whenever a relatively large amount of historical data exists for the APT method at all relevant laboratories, further risk-based elimination(s) could be considered based on a risk-benefit analysis. In another example, if the APT method has been used for multiple products and appropriate test system controls exist, the APT method could be verified (“validated”) at the sending laboratory without repeating this later at the receiving laboratory. This would essentially remove the typical AMV stages 2 and 3 and would significantly reduce time and resources. The validation master plan (VMP) is typically used to capture these reduction strategies.
Table II lists typical DS test methods and relevant specification examples for a monoclonal antibody (mAb). As highlighted in green in this table, extensive opportunities exist to develop and use APT test methods. The use of APT methods goes beyond mAbs, and the APT concept can also be used for other platform product types such as messenger ribonucleic acid (mRNA)-technology-based vaccines, antibody-drug conjugates (ADCs), and so forth. The APT concept does however not apply wherever test methods are specifically developed for a new product. For example, potency tests that are ideally based on the mode-of-action (MoA), simulating the in vivo biological activity, are not suitable for APT.
Typical Late-Stage DS Specification Tests and Specifications (Intended Use)
The specification examples provided here are intended for late-stage IMP manufacturing, and narrow the intended use for the test method(s) to be validated. Conceptually, the preferred pathway is to use these product-specific late-stage DS and DP specifications as a primary driver for setting AMV protocol acceptance criteria. This applies to the initial AMV studies and well as all subsequent APT verification studies (8).
For example, in the high-performance size-exclusion chromatography (HPSEC) separation method, the purity specification of no less than (NLT) 98.3% drives the acceptance criteria for “suitable” method performance in an “outside-in” approach. The term “outside-in” simply means working inwards for acceptable method performance levels from the outermost relevant reference point (specification(s)) (8). The maximum total error allowed in the AMV studies does not exceed a specified and justified portion of the difference(s) between the specifications and the representative DS/DP release/stability data. As the test method is validated and deemed to be suitable, it can reliably and accurately deliver test results within and outside of the specification range.
5. HPSEC Case Study
In this case study, we will use the HPSEC test method, because it is a common separation test for biologics. It is used as a primary method to control impurity and purity levels. The case study has essentially two parts and starts with the preparation and execution of the initial AMV study and the following AMM program. The second part illustrates the suggested APT verification studies.
5.1. Initial AMV Studies and AMM Program
Regardless of whether a pre-AMV AMT study is required, the robustness study is completed. At this point, we are technically still in AMV stage 1 (qualified method status). Using the late-stage DS (and DP) HPSEC specifications from Table II (i.e., Major product peak: NLT 98.3%; Aggregates: No more than (NMT) 1.7%; Fragments: NMT 1.7%) to frame the suitability of use for this method, we can evaluate the readiness of the AMV and prepare the AMV protocol for this quantitative limit test.
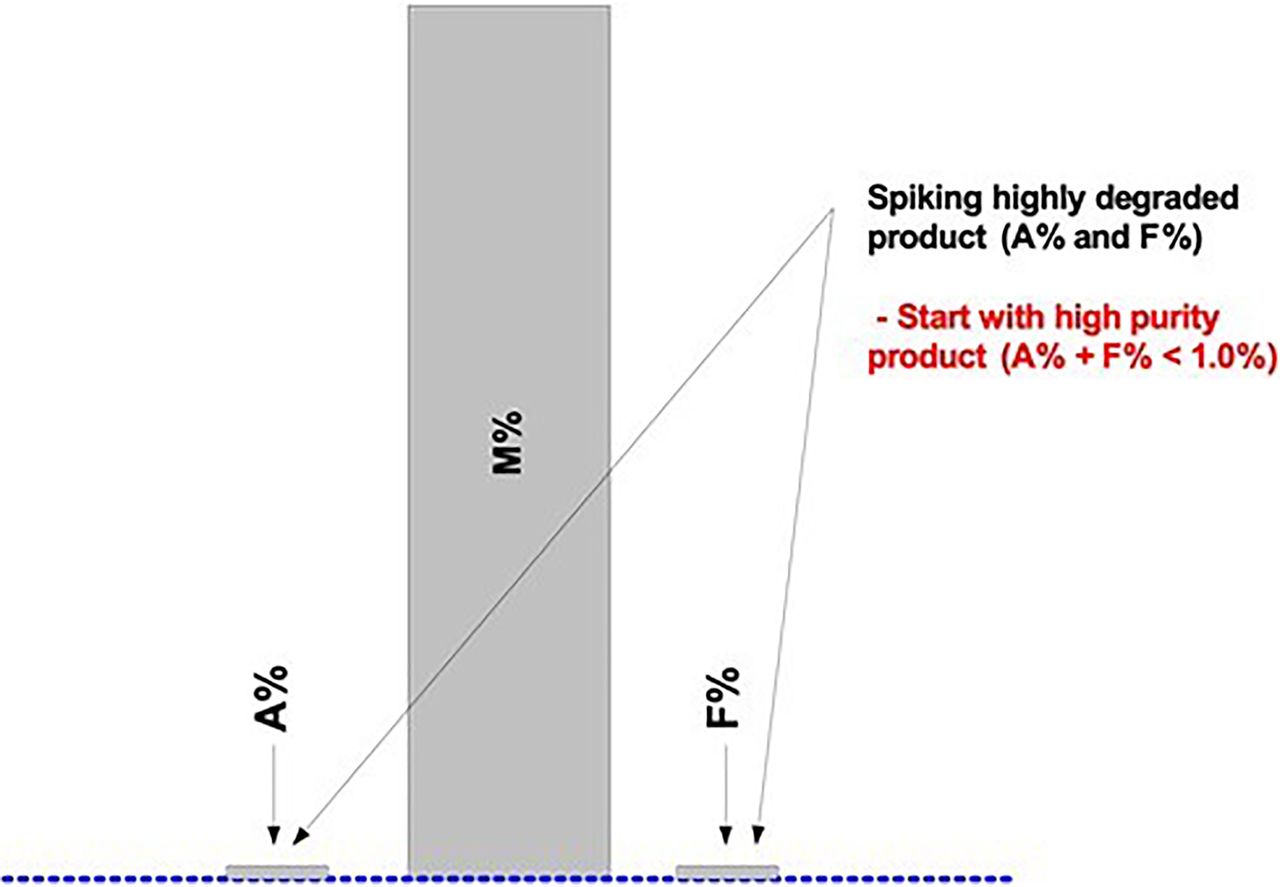
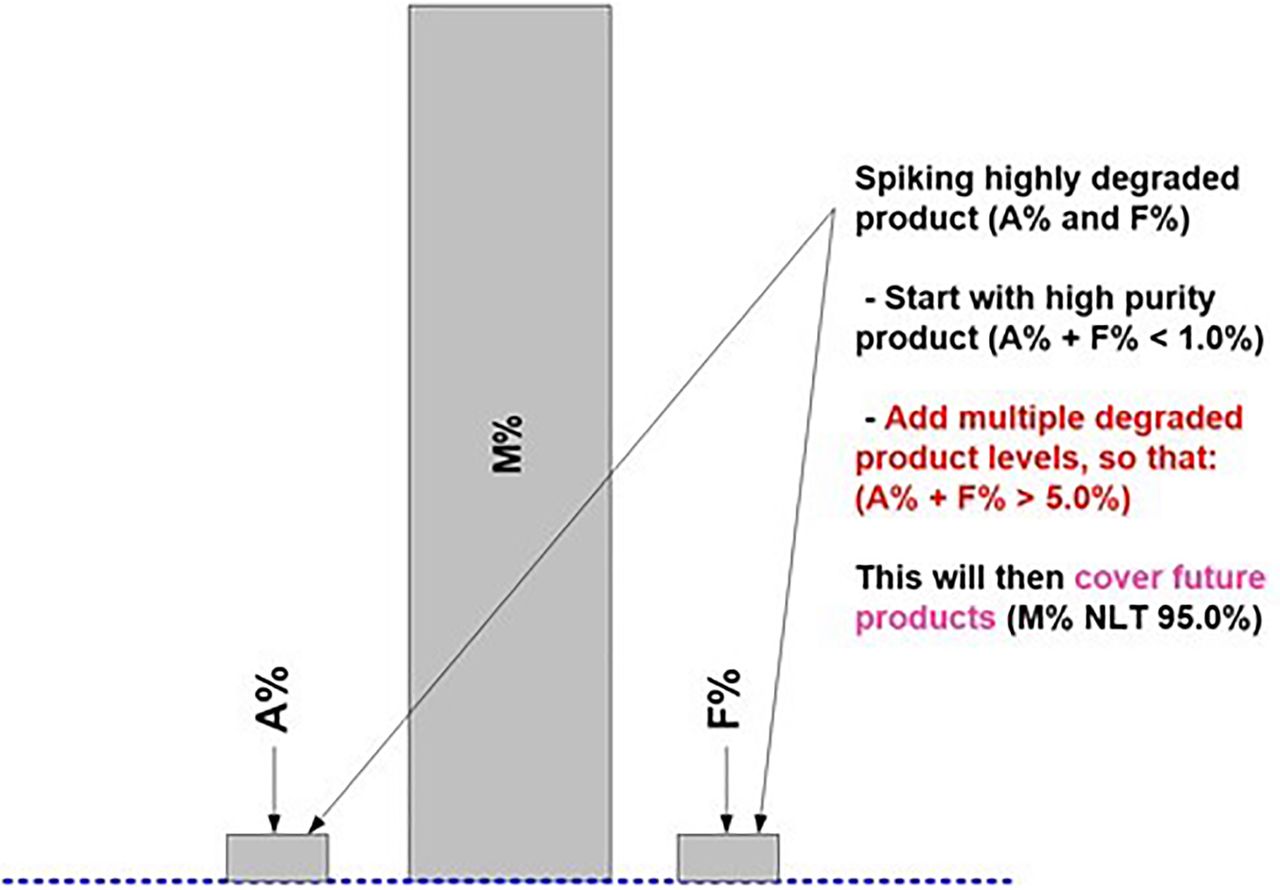
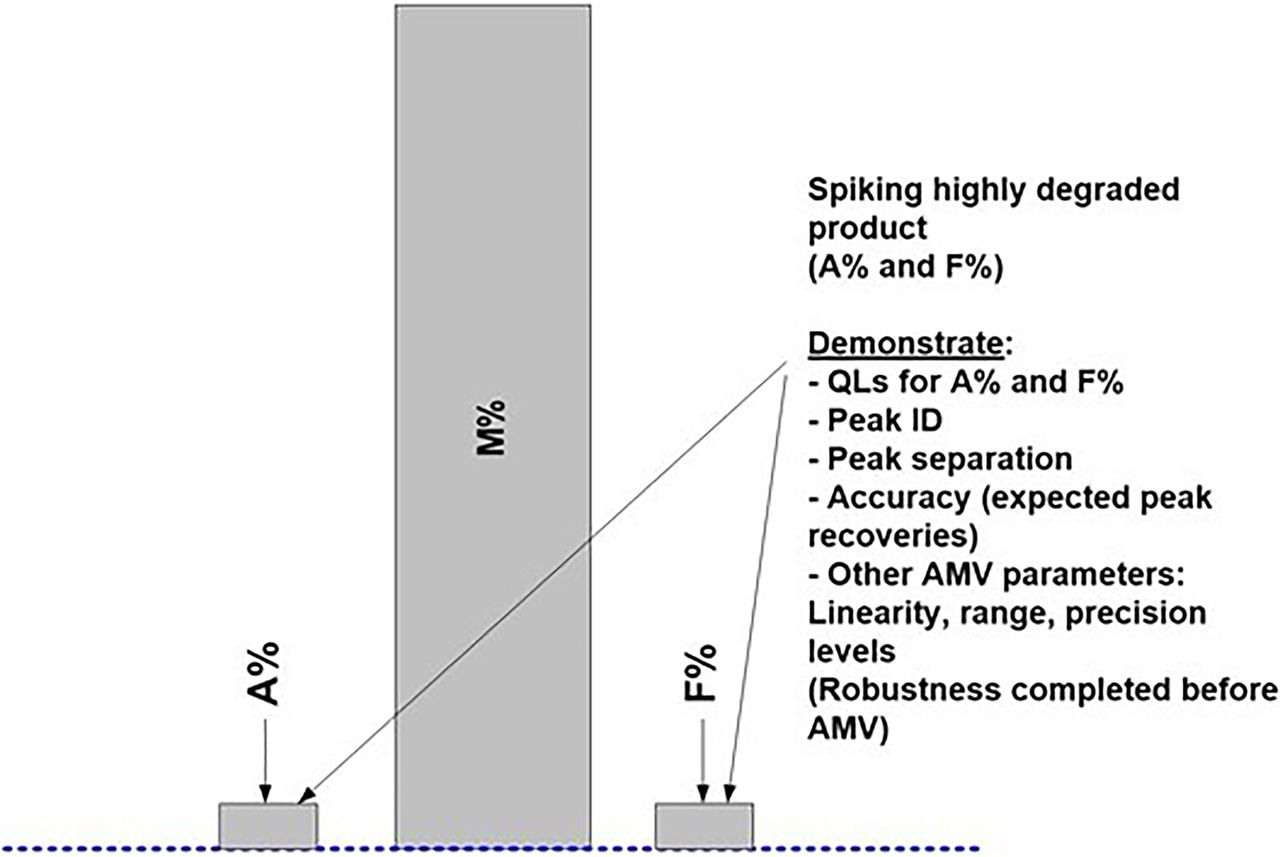
As part of the method readiness evaluation, we also assessed the additional method characteristics to be considered before AMV study execution or during the AMV study. Table III lists the most relevant additional method characteristics to be considered. Figures 7⇓–10 are used to illustrate the required spiking and supporting studies to validate all potential test results (Impurity A = Aggregates; Impurity F = Fragments; Purity M = Monomer) from the lower QL impurity level(s) to beyond the DS/DP specification level(s).

AMV study example(s) purity by HPSEC—initial AMV study.
AMV, analytical method validation; HPSEC, high-performance size-exclusion chromatography.

Initial AMV study (continued).
AMV, analytical method validation; NLT, not less than.

Initial AMV study—for all validation characteristics.
AMV, analytical method validation; QL, quantitation limit.

APT method AMV study.
AMV, analytical method validation; APT, analytical platform technology; QL, quantitation limit.
Supporting AMV Method Performance Characteristics for HPSEC (Ideally pre-AMV)
Figure 7 represents a typical, high-purity DS/DP product chromatogram with relatively low levels of Impurities ([A + F] < 1.0%). In this example, Impurities A and F are spiked using highly degraded levels for each impurity (Figure 8). The purity (M) is not directly manipulated to achieve the desired bracketing of the specification level(s). Any or both of the spiked (or blended) impurities will proportionally lower the purity level (M = 100.0% − [A% + F%]). A representative DS/DP lot with very low impurities should be used for the spiking studies to achieve the validation of a relatively low QL (Figure 7).
When using the ICH Q2(R1) approach “C”, which is based on a linear regression analysis of the added spike levels, or any other justified alternative approach, several points should be considered (9). Replicates of each spike level used as individual results (instead of averages) may yield higher standard deviations (SDs) and therefore higher QLs. Approach C may provide low QLs when the assay response is highly linear, precise, and accurate over the selected range. Spiked sample preparations should be accurate and precise to prevent random and systematic deviations from the regression line, as they may increase the QL. Excluding higher spike levels, if possible, usually results in a relatively low QL determination. Potential outliers in higher spike levels may significantly increase the QL due to an increased “leverage effect” (8).
It is further suggested to consider stretching the desired bracketing range(s) of impurity levels far beyond initially required (product-specific) AMV levels (Figure 8). This will then likely validate any additional similar product regardless of the product-specific impurity levels and/or life cycle stage(s).
Some of the remaining validation characteristics can be taken and summarized from the initial AMV studies. Ideally, method robustness studies were completed prior to the initial AMV studies. Any required operational conditions were adjusted/tightened as suggested by the robustness study results. Other method performance attributes such as frequency of invalid tests, peak separation, integration consistency, and stability-indicating capability can often be used from existing, routine method performance trending data acquired before the AMV study.
Once the AMV study is completed with passing acceptance criteria and any execution exceptions resolved, the AMM program starts. The control of long-term analytical method performance is critical to an APT. A rigorous change control program that carefully evaluates and qualifies method element changes such as changes in reagents, instrumentation, chromatography software, and so forth should be used. The demonstration of long-term method control will be key to a regulatory approval as an APT.
When establishing the AMM program for the APT method, it should be recognized that the use of a nonproduct-specific assay control can greatly simplify system control and routine laboratory operations. For example, for mAbs, the National Institute of Standards and Technology (NIST) mAb IgG1k monoclonal antibody reference material could be a suitable assay control (11). Or, a representative in-house mAb manufacturing lot can be used. Using an assay control with low impurity levels may not monitor the method performance in the critical range close to the out-of-specification (OOS) level(s). Higher than typical impurity or degradation levels may provide better system suitability control and will provide more confidence in the test results when results are close to the OOS level.
In this last AMV life cycle step, the system suitability of the test method is closely monitored and periodically adjusted to a desired level of system control. Although AMV study results for intermediate precision are often used to represent the performance capability of the test method, it is the trending data from the system suitability controls that are more representative of its long-term capability. Ideally, the AMV data from this long-term life cycle step should be used to estimate the contribution of all relevant manufacturing and product stability data to the variability of the analytical method. This is conceptually similar to process validation stages 2 and 3, in which the manufacturing control limits for CPV are established and adjusted from a long-term manufacturing data evaluation (e.g., n = 15+ lots) instead of using only a subset of available data (e.g., n = 3 lots from PV stage 2).
5.2. APT Verification Studies
Once the test method is agency-approved via the marketing authorization, this test method is a potential APT method, but supporting data from other relevant products, demonstrating the ability of the method to be an APT, should be provided in subsequent applications. Upon approval of the subsequent product, the test method is considered an approved APT method. If APT is being claimed and a sponsor is seeking authorization for the method to be approved as an APT in a market authorization, application data should be provided to support the claim. This data should demonstrate that the method meets the definition of an APT, which is an analytical method that can be used for multiple products or different sample matrices without modification of the procedure. Supporting data from at least one additional product should be provided to support the APT.
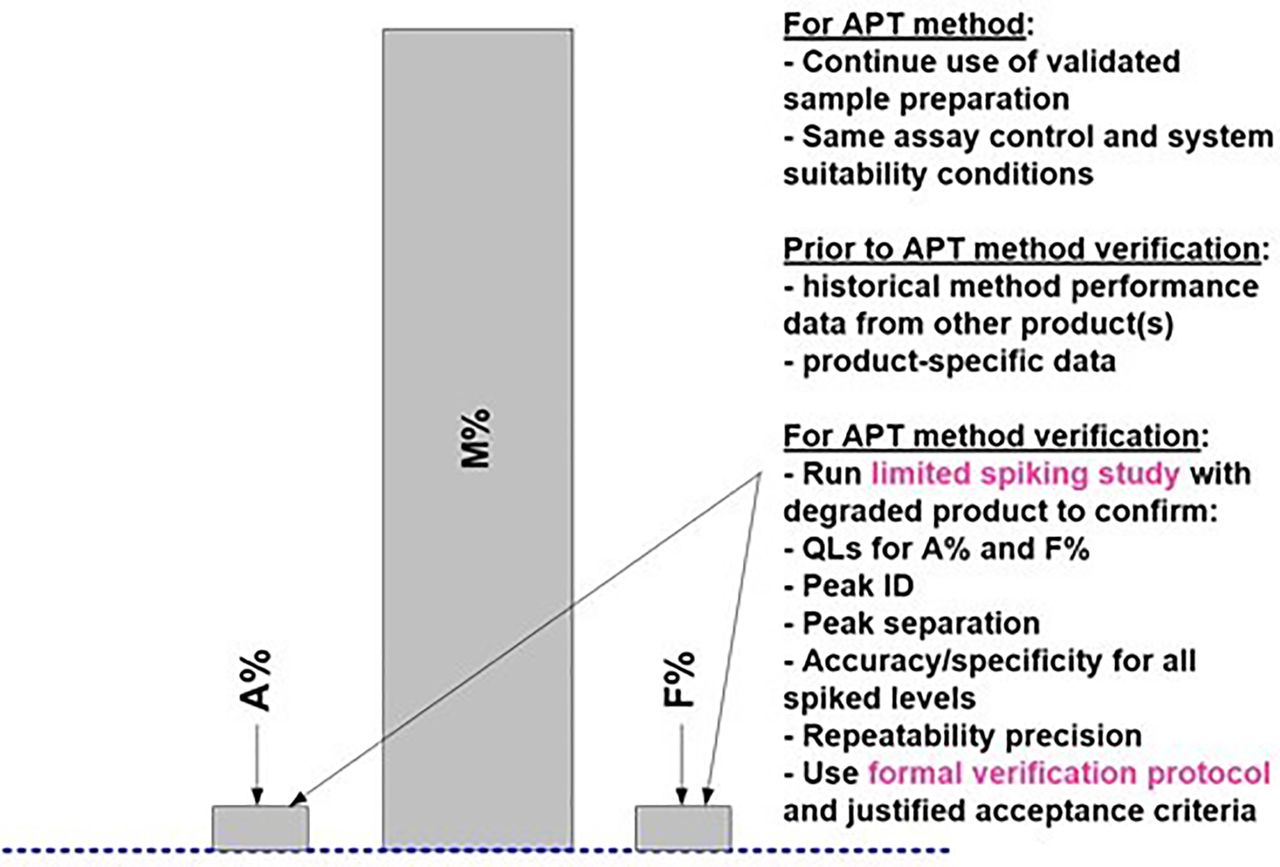
Once an APT method is established, for any similar new product starting IMP manufacturing, reduced prospective APT verification studies can be formally used instead of a full set of AMV studies. Figure 10 and Table IV capture the suggested prospective APT verification studies for the HPSEC case study. As mentioned earlier, AMV-stage verification studies will be similar to AMQ-stage studies. There is an option to use the initial validation study results for some of the proposed validation characteristics (Table IV) beyond those suggested, and thus not repeat them for the APT verification studies. However, some prospective verification studies, covering accuracy and specificity (and QL if needed), are still functionally required for “approved” methods for this quantitative limit test. As captured in Figure 10, the primary intent of the APT verification study is to confirm a suitable accuracy and specificity method performance for the new product. In addition, a suitable QL is also confirmed by using an appropriate spiking series for the accuracy study. The suggested set of spiked samples provides all relevant method performance characteristics results, is relatively easy to prepare, and can be executed essentially in one day as shown in Table IV. Other test method performance characteristics, such as robustness and intermediate precision, which typically take an extensive amount of laboratory work and time, are not required to be repeated.
Prospective APT Verification Study Design for HPSEC Method
The prospective study results should be combined in the APT verification report with relevant retrospective results (initial AMV study and/or AMM results), as illustrated in Table V. The same set of data from the prospective spiking study to confirm suitable accuracy is also used to confirm specificity, linearity, assay range, and QL. As mentioned earlier, the other resource/time-consuming test method performance characteristics are not repeated, but relevant results from the initial AMV studies (and AMM data as relevant) are captured in the APT study report. Documenting the initial AMV and AMM method performance data/results in the APT report has the advantage of having available a single report supporting the marketing authorization.
Combined Retrospective and Prospective APT Validation Results
Further eliminating prospective APT method validation characteristics should only be considered when the new DS/DP product and the product tested in the initial AMV study have very similar separation profiles and impurity levels. For the typical separation test methods, which can be used and verified as APT methods, the product-specific elution profiles and impurity levels may greatly impact the QL determination or verification.
For the marketing authorization submission, the sponsor should submit the initial AMV study report and product-specific verification results and provide a summary table containing all relevant initial AMV and APT verification results. CDER/FDA recommended to clearly justify the use of the APT in each BLA, and to provide rationale why a particular product is suitable for APT use. For example, the rationale could contain information on product similarity (i.e., similar to previous product). For the completeness of relevant information contained in the APT verification report, the following retrospective data should be included:
All retrospective AMV study results and acceptance criteria not repeated in the APT study.
The long-term AMM assay control performance (% CV) can be used alternatively or in combination with AMV Intermediate Precision.
Pre-AMV supporting method attributes (Table III) as relevant.
When using an APT method for another product, it is important to review the method change log(s) and to confirm that no significant changes to the test system have occurred. Before completing all APT verification/validation steps (AMQ, AMT, AMV), it should also be confirmed that the AMV study results (e.g., QL(s)) are suitable for the next product. Another consideration is the option to use the initial AMV study acceptance criteria, if relevant, for the APT verification study protocol. Depending on the DS/DP specifications of the new product and the relevant manufacturing capability, they may no longer be suitable for a new product.
6. Discussion
The use of the APT verification concept can be applied to new products regardless of whether they are in an accelerated development or not. The value for the development organization is greater for accelerated development as available time and internal resources become tighter. The strength of the APT concept lies in the shifting of some of the traditional laboratory work toward a risk-based documentation exercise. Although this will greatly help laboratory operations and the supporting functions to focus on other tasks, the APT “readiness” review process and overall validation report package assembly may require careful planning. A deep conceptual understanding is essential to plan how product-specific prospective study results should be added to relevant retrospective AMV results.
The APT concept can also be applied to product and process characterization tests that typically are only qualified. Although characterization tests are not “approved” by the agency, a similar AMQ concept can be applied.
In the 2015 CDER presentation, the author proposed several BLA submission options (5). The options (A–C) received the following comments from CDER:
Option A: The sponsor submits only the results from APT method verification studies. The test method is described in the license application, and the reference is provided to the relevant section in previously approved BLA. The initial AMV results are not provided. CDER did not consider this an acceptable option as each BLA is reviewed as a stand-alone submission.
Option B: The sponsor submits initial AMV study acceptance criteria and results in a summary table format plus product-specific APT verification acceptance criteria. The summary table distinguishes results from the initial AMV study and product-specific APT verification data. CDER much preferred this option over option A and indicated that this could be acceptable if the suitability of using APT verification data can be fully justified for a specific test method.
Option C: The sponsor submits initial AMV study report and product-specific verification results and provides a summary table containing all relevant initial AMV and APT verification results. CDER considered this to be the best option, because CMC reviewers may not have insight into the previous BLA. There are many reviewers, and data must be available to make a recommendation for approval. CDER recommended to clearly justify the use of APT in each BLA and to provide rationale why a particular product is suitable for APT use. For example, the rationale could contain information on product similarity (i.e., similar to previous product).
CDER indicated that the agency welcomes the use of the APT concept in future submissions.
7. Conclusion
Using an APT concept can significantly support accelerated product development by reducing prospective laboratory testing typically performed for method qualification, transfer, and validation. As illustrated in the HPSEC case study, the initial AMV (stage 2) and AMM (stage 3) study conditions and results become the foundation for the much-reduced prospective APT verification studies and reports.
A strong foundation is required for the successful use and approval by the agencies. This is accomplished by practically converting a line-of-sight vision toward future APT use for the analytical development and validation studies and when demonstrating a continuous controlled and validated state. When submitting APT verification study results in marketing applications, the sponsor should consider submitting rationale for APT suitability and all relevant initial AMV study conditions and results and all relevant evidence for continuously controlled AMM state for this APT method.
Conflict of Interest Declaration
No financial or nonfinancial competing interests exist related to the manuscript.
Acknowledgement
The author is very grateful to Dr. Marjorie Shapiro, OBP/OPQ/CDER/FDA, for her thoughtful comments on an earlier draft version of this manuscript. The author is also grateful for the many helpful comments received on a later draft version by Dr. Jayda Siggers, CERB, BRDD, HPFB, Health Canada, as well as Jane Halpern, Consultant, and Dr. Earl Zablackis, Sanofi.
Footnotes
↵i The content and views expressed in this article by the author are not necessarily views of the organization he represents.
- © PDA, Inc. 2022




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}