
Abstract
When an initial marketing authorization of a pharmaceutical product is granted, a substantial number of chemistry, manufacturing, and control (CMC) post approval changes (PACs) have to be managed by the manufacturers. Despite efforts undertaken over the years by multiple regulatory jurisdictions, there is still heterogeneity in terms of regulatory requirements and timelines across national regulatory authorities (NRAs). This creates complexity in managing global CMC PACs, putting the supply of medical products at risk. Regulators have developed regulatory mechanisms that aim at accelerating the reviews and approvals of PACs by NRAs. The World Health Organization (WHO) is supporting the concept of “reliance” among NRAs, which are encouraged to rely on the assessment completed by a “highly performing authority”. The objective is to accelerate the overall process for PACs, ultimately fostering more equitable and timely access to medical products for populations who need them. With the support of Health Canada, WHO, Pan American Health Organization, and the Paul-Ehrlich-Institut, Sanofi has launched a pilot using the principles of reliance for a CMC PAC for a vaccine, with 21 NRAs who accepted to participate in the pilot. The objective of this pilot was to apply these principles to reduce the approval timeline to a maximum of 6 months in all countries after an initial approval is granted by a reference authority. We discuss the opportunities and challenges of implementing reliance principles for CMC PACs. We also describe the pilot experience by sharing initial lessons learned from the Step 1 of this pilot, which consisted of engaging the reference authority and the NRAs.
Introduction
Initial marketing authorizations for medical products are only the first step in making products available. Significant chemistry, manufacturing, and control (CMC) post approval changes (PACs) occur to ensure a continuous supply of high-quality medical products in a cost-efficient way. Dellepiane et al. (1) have identified four main categories of PACs for vaccines: 1) routine changes needed to ensure continuous manufacturing and control activities (e.g., new working seed lot to replace the previous one, which is depleted), 2) changes performed in the frame of continuous improvements (e.g., quality control method improvement), 3) changes to ensure supply continuity and meet supply demand (e.g., new manufacturing site), and 4) changes to introduce innovations (e.g., replacement of in vivo by in vitro testing).
Although CMC PACs apply to all types of medical products, PACs associated with vaccines can be some of the most complex to manage. This is because the manufacturing of most vaccines is complex owing to the involvement of a high number of raw materials, manufacturing steps, and pieces of equipment and to requirements for a high number of quality control tests and the critical reagents they require. Different vaccines use different antigens, so one PAC in an antigen can impact multiple final drug products, such as combination vaccines. Moreover, the overall time required to manufacture and control vaccines can be up to 3 years from the first production stage until the release of a batch.
Regulations across the world are mostly nationally based and still very heterogenous (1⇓⇓⇓⇓–6). On the global stage, manufacturers have to face different legislative frameworks, regulatory requirements, regulatory procedures, timelines (which can be unpredictable), resource levels to manage scientific and regulatory work, and level of maturity of national regulatory authorities (NRAs). Numerous efforts have been undertaken by international institutions to move toward greater harmonization of procedures and requirements. The World Health Organization (WHO) certification scheme was set up in 1969, with the goal of enabling countries with limited drug regulatory capacity to obtain assurance that the pharmaceutical products to be imported are of good quality, safe, and effective (7). Although WHO does not publish the list of countries involved in the scheme, the 1997 report indicates that, at that time, a total of 141 countries had expressed their intention to be a part of the scheme. In parallel, in 1993, the European Medicines Agency (EMA) introduced a centralized regulatory review procedure for enabling manufacturers to submit a single market authorization application in the European economic area applicable to all member states for certain medical products (8). This procedure applied to initial applications and then subsequently for any PACs requiring regulatory review and approval. This framework allows an efficient and secure pathway that ensures regulatory oversight and introduces consistent requirements while eliminating nonvalue-added duplication using principles of reliance.
The International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) issues international technical guidelines that every member is expected to adopt and transpose into its national regulation. ICH is composed of 14 members and 22 observers from national authorities (9), that is roughly 19% of the 193 United Nations members. Pharmaceutical Inspection Convention/Cooperation Scheme (PIC/S) is a nonbinding, informal co-operative arrangement between regulatory authorities in the field of good manufacturing practices (GMPs) of medical products (10). It aims at harmonizing inspection procedures worldwide by developing common standards in the field of GMP. The PIC/S is composed of 54 members, that is, representing 28% of the United Nations members. International Pharmaceutical Regulators Program (IPRP) was created in 2018, with a purpose of creating an environment for its regulatory members and observers to exchange information on issues of mutual interest, enabling cooperation and promoting convergence of regulatory approaches for pharmaceutical medicinal products for human use (11). The WHO issues guidelines to help NRAs strengthen their regulatory systems. However, it is up to each NRA to decide on the adoption of those guidelines, as WHO has no enforcement power. Although these efforts have attempted to introduce consistency, this has yet to fully materialize. Because of this heterogeneity, a PAC must be submitted to multiple NRAs, following national requirements and timelines, and in some cases requiring up to 4 years to achieve approval in all targeted countries. Considering that it is not possible for vaccines manufacturers to handle multiple versions of a manufacturing process or quality control method while waiting for approvals from multiple countries, this situation creates tremendous complexity. Several national regulations require industrial implementation of the change within a specified timeline after their national approval. Implementation of a PAC before approval has been obtained in all targeted countries may be done, for instance, when most countries have approved the PAC, or when the PAC is necessary for the continuous manufacture and supply of the product. However, this approach may present a supply continuity risk for patients in countries where the PAC has not yet been approved.
The International Federation of Pharmaceutical Manufacturers and Associations (IFPMA) discussed this situation in its infographic “The complex journey of a vaccine”, suggesting that about half of the world population is at risk of supply shortage for vaccines, in part as a result of this complexity (12).
The need to significantly improve this situation has been recognized by ICH members, as reflected in the introduction of the ICH Q12 guideline “Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management” published in November 2019 (13). However, more than 3 years after this publication, few ICH members have started implementing some of the principles outlined in this guideline. Similarly, in 2015, the WHO issued “Guidelines on Procedures and Data Requirements for Changes to Approved Vaccines” (14). It provided guidance to NRAs for harmonizing their regulatory processes with regards to the management of PACs. Again, there are still many NRAs that have yet to implement the principles described in this guideline 7 years after it was published. This delay is likely due to the need for NRAs to revise their national regulations, which is typically a lengthy process.
More recently, in 2021, the WHO issued the Good Reliance Practices (GRelP) guidelines (15). These guidelines aim at promoting a more efficient approach to regulation, thereby improving access to quality-assured, effective, and safe medical products. The document presents the overarching principles of regulatory reliance in the oversight of medical products and the use of reliance as a lever to improve the efficiency of regulatory oversight. In addition, the International Coalition of Medicines Regulatory Agencies (ICMRA) decided to leverage lessons learned from the COVID-19 pandemic by launching pilots on a collaborative assessment of PACs (16). This consists of using “a collaborative assessment framework to identify and exploit areas of alignment between different regulatory agencies in terms of quality assessment for PACs”. Initiatives such as the ORBIS project (17), the ACCESS consortium (18), and the OPEN initiative (19) are other initiatives aimed at developing collaboration across multiple NRAs. Several health authorities in the world have also decided to work collaboratively through multilateral agreements as described by Gastineau (6).
These initiatives are steps moving in the direction of global convergence, harmonization, collaboration, and reliance across countries. This is aligned with some of the pharmaceutical industry requests, as reflected by Deavin et al. (20) in the position paper on “Path Forward to Optimise Post-Approval Change Management and Facilitate Continuous Supply of Medicines and Vaccines of High Quality Worldwide” outlining the European Federation of Pharmaceutical Industries and Associations (EFPIA), Vaccines Europe, and IFPMA joint position. Illustrations of what trade associations are advocating for have subsequently been published under the Pink Sheet (21).
In addition, the COVID-19 pandemic demonstrated that it was indeed possible, under a public health emergency, to overcome some of the regulatory barriers by finding ways to grant approvals and enable fast access to medicines and vaccines. WHO has been able to grant an emergency use listing (EUL) status to some vaccines against COVID-19 in record time by relying upon the review and approval of a stringent regulatory authority used as a reference. In turn, the EUL facilitated in-country authorizations of the COVID-19 vaccines by using reliance mechanisms (22). The collaborative registration procedure that WHO has set up is also a great way to enable more collaboration across NRAs. Its objective is to accelerate the registration of medical products across NRAs by leveraging assessment and inspection outputs already produced by WHO prequalification (23).
Although collaborative processes are being tested, it does not mean that the NRAs involved in a collaboration will rely upon approvals granted by the NRAs who performed the review. Also, although reliance principles have been set up, the way to translate those principles into concrete actions has not been widely and globally accepted, specifically for the management of CMC PACs.
In order to test the principles developed under the GRelP and generate a broad understanding of both opportunities and potential obstacles, Sanofi has launched a pilot testing of regulatory reliance principles for a vaccine CMC PAC. Although subsets of preset countries or regions are testing reliance principles, the countries identified for this pilot are driven by the those impacted by this variation. In this case, we are testing the flexibility and scalability of using the reliance principle for multiple different groups of countries driven by the PAC. This article describes the approach taken and outlines the lessons learned from the first of the two steps of this pilot.
Methods
The following section describes how the test case was identified and the parameters of the pilot.
Case Study Selection
The selection of the CMC PAC case study was based on the following criteria: 1) the intended PAC was categorized as requiring a regulatory approval by the country of origin before the release of batches impacted by the change; 2) it was a low complexity PAC, considering the typical scientific data to support the change; 3) the intended PAC fell within the PAC categories outlined in Dellepiane et al. (1), that is, manufacturing site transfers; 4) the PAC’s timing allowed sufficient time to engage in preliminary discussions with stakeholders and to suggest a reliance approach achievable by most countries without any time constraints; 5) the PAC was representative of real conditions (i.e., impacting numerous countries in different parts of the world); and 6) the reference regulatory authority was willing to participate and support the experiment.
Based on these criteria, the PAC selected was the transfer of the filling and packaging activities of a single-dose vial presentation for a combination booster vaccine from a Sanofi manufacturing site in Canada to another Sanofi manufacturing site in France. The vaccine is WHO prequalified.
Some minor changes to the manufacturing process had to be considered at the same time, because it is a rare occurrence to transfer manufacturing activities from one site to another under strictly identical conditions. In addition, the official release of the batches was also to be transferred from the Canadian national control laboratory to the German Paul-Ehrlich-Institut (PEI) laboratory.
Countries Impacted
This single-dose vial vaccine is currently licensed in 40 countries around the world, with the first license granted in 1999 and the most recent in 2021.
At the time of considering this case study, Belarus, Kazakhstan, and Russia were classified as out of scope, as the submission of the PAC could not be managed while the licenses in these countries were being converted under the EurAsian Economic Union mutual recognition procedure. This left 37 countries spread over 4 continents in the scope, including 2 countries in the EU where the vaccine is authorized under a mutual recognition procedure, Canada as the reference country, and the WHO, which had granted the WHO prequalification status.
Overall Objective
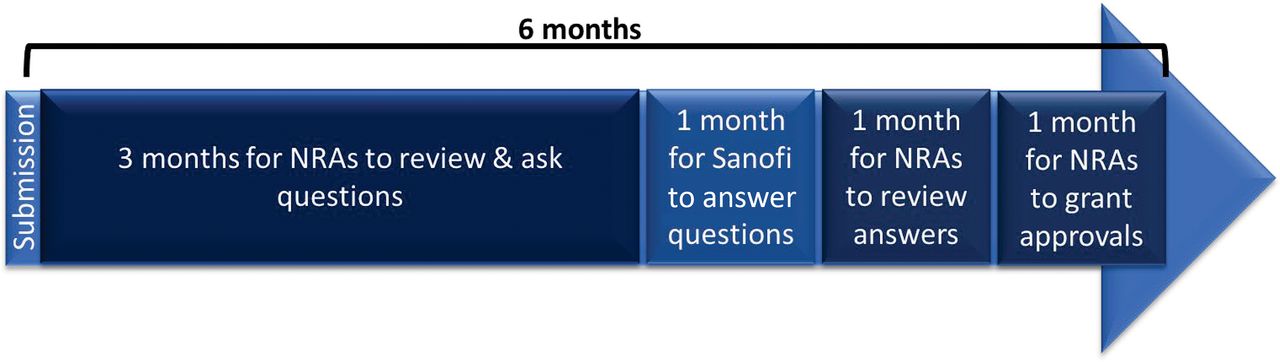
Based on past experiences, the review and approval of the above type of change by such a number of countries were expected to take between 6 months and 4 years. This pilot was set up to test convergence in assessments and timing and reliance principles, with the overall objective of having the PAC reviewed and approved in all participating countries within 6 months after submission, as per the WHO guideline on PACs of vaccines (14). The overall approach consisted of 2 main steps:
Step 1: Submit a dossier and receive approval from Health Canada's Biologic and Radiopharmaceutical Drugs Directorate (BRDD) as the responsible NRA for this impacted WHO prequalified vaccine
Step 2: Submit the same dossier (including the questions asked by the BRDD and responses provided by the company) to all impacted NRAs and WHO for review, relying upon the initial assessment of the BRDD. To encourage NRAs and WHO to apply reliance, the company shared the unredacted assessment report from the BRDD. It was made clear that the relying agency retains the final decision of whether to approve the variation or not.
Overall Process and Timing
The process and timing for Step 1 was dictated by Health Canada’s regulations for a prior approval type of application, that is, a maximum 6-month review and approval period (after a 45-days screening phase), including questions and answers. The 6-month timeframe for Step 2 was proposed by the company, as depicted in Figure 1.

Proposed timelines for the review process of the pilot.
Success Factors
The following success factors had been selected before launching the pilot:
Support from Stakeholders:
As this was a pilot, the company considered that the support from a number of key stakeholders was an important aspect to obtain participation from as many NRAs as possible. First, Health Canada, a “highly performing NRA” (NRAs exhibiting a high level of performance of WHO’s six recommended regulatory functions and exercising full regulatory oversight of any given vaccine) (24) and the responsible NRA for this WHO prequalified vaccine, was asked to share its assessment report with other NRAs. Next, the German authority (the PEI), also a “highly performing NRA”, was involved for being the future reference authority, the receiving country of the future official testing and release activities, and the potential reference member state in the EU for the mutual recognition procedure applied for this vaccine. Also, the involvement of WHO was important as the vaccine is WHO prequalified and the organization is strongly supporting the use of reliance mechanisms. Last, support from the Pan American Health Organization (PAHO) was essential because 46% of the countries in scope of the pilot were from the Latin America.
Product Sameness:
Ensuring product sameness across NRAs is an important prerequisite to apply reliance mechanisms so that NRAs can assess the application based on the assessment performed by another authority. Product sameness is defined in the WHO GReIP as the product being submitted to the relying authority and the product approved by the reference regulatory authority should be essentially the same (15). IFPMA has also issued a position paper providing an industry-focused interpretation of this terminology (25). In this case study, the vaccine manufactured is differentiated at the packaging step and is therefore the same in all countries, based on a “one size fits all” principle. This principle is an important element to optimize the use of resources, unless otherwise justified.
In an effort to optimize resources, the pilot identified the following six requirements. First, the exact same dossier was submitted to all participating NRAs, apart from the administrative local information as per the local legal obligations. In other words, all regulators receive the same data. This was based on a pre-evaluation by the company of the specific technical requirements in some countries. Moreover, requirements not considered as scientifically justified were not to be met by the company. Second, the unredacted assessment report from Health Canada was submitted to all participating NRAs. Third, all questions (if any) and answers were consolidated into a single document (facilitated by a digital tool) submitted to all participating NRAs. Fourth, the same process and timing were to be followed by all participants. Fifth, there was no GMP inspection of the French receiving site, as it was already regularly inspected by the French authorities. Last, no product testing by NRAs was to be required in the frame of this submission, as it was not justified for this type of change according to the Canadian regulations.
Participation in the pilot required agreement to these conditions to respect the components of the pilot and to ensure the sameness of criteria across all Agencies.
Transparency:
Transparency is a prerequisite for building trust which, in turn, is a key success factor for applying reliance principles. This was illustrated by the company and stakeholders, by sharing the unredacted assessment report (as agreed with Health Canada) and all questions and answers—consolidated in a single document—to all the participating NRAs. No additional documentation was required of participating Agencies to receive the unredacted assessment report or access to the Q&As.
Digital Tool:
In order to facilitate the sharing of questions and answers in real time, the company has developed a simple and intuitive proof-of-concept tool that enables all reviewers to access and sort all the questions raised and the answers provided during the assessment phase.
Results
Encouraging participation in the pilot as well as understanding the barriers for participation were key factors and considerable efforts were made to support this objective.
Pilot Preparation
The pilot preparation consisted of creating an internal cross-functional team, with representatives from regulatory affairs, public affairs, and quality departments. Two main tasks were defined and undertaken. To start with, discussions were initiated with the four external stakeholders (Health Canada, WHO, PAHO, and German PEI) to get their buy-in and support. Then, discussions were held with Sanofi regulatory affiliates in all 37 impacted countries, first, to understand the local specific requirements and identify potential issues and, second, to engage local discussions with all NRAs to get their agreement for participating.
The preparation for this pilot started in 2019. Although it was initially considered as very early before the target dossier submission date to Health Canada in April 2022, and then to all NRAs and WHO in early 2023, this first phase required significant planning and dialogue both within the company and with external stakeholders. This included engaging with multiple company country affiliates and corresponding agencies, which had considerable and sometime conflicting priorities, including the COVID-19 pandemic. It also required presenting different ways of working, getting agreement, and addressing perceived barriers.
Liaising and Meeting with Stakeholders
The company first engaged discussions with each of the four external stakeholders individually to ensure support. All stakeholders immediately agreed to participate and to support the pilot. Once this was completed, the company organized regular joint meetings with all four stakeholders. The COVID-19 pandemic impacted the company's interactions with NRAs; however, the momentum was maintained—thanks to the stakeholders’ support. Even in a pandemic situation, the stakeholders expressed their support for the pilot in videos that were shared with the NRAs. From April 2021 to December 2022, Sanofi country affiliates contacted the NRAs to present the pilot either by e-mail, via local regulatory platform, or during meetings. In the end, 21 countries agreed to participate with the final 4 joining as a result of efforts by WHO and PAHO during conferences.
As part of the pilot to utilize a standardized dossier, the company asked Health Canada to adjust their dossier requirements with regards to the Module 3 – Facilities section (3.2.A) of the dossier by adopting the EU requirements as the standard. The information usually required by Health Canada is much more detailed than that in the EU, which manages this information under the GMP regulations rather than the marketing authorizations. Health Canada agreed with the proposal by taking a pragmatic approach consistent with the level of expectations on that aspect from other NRAs in the world.
Participating NRAs
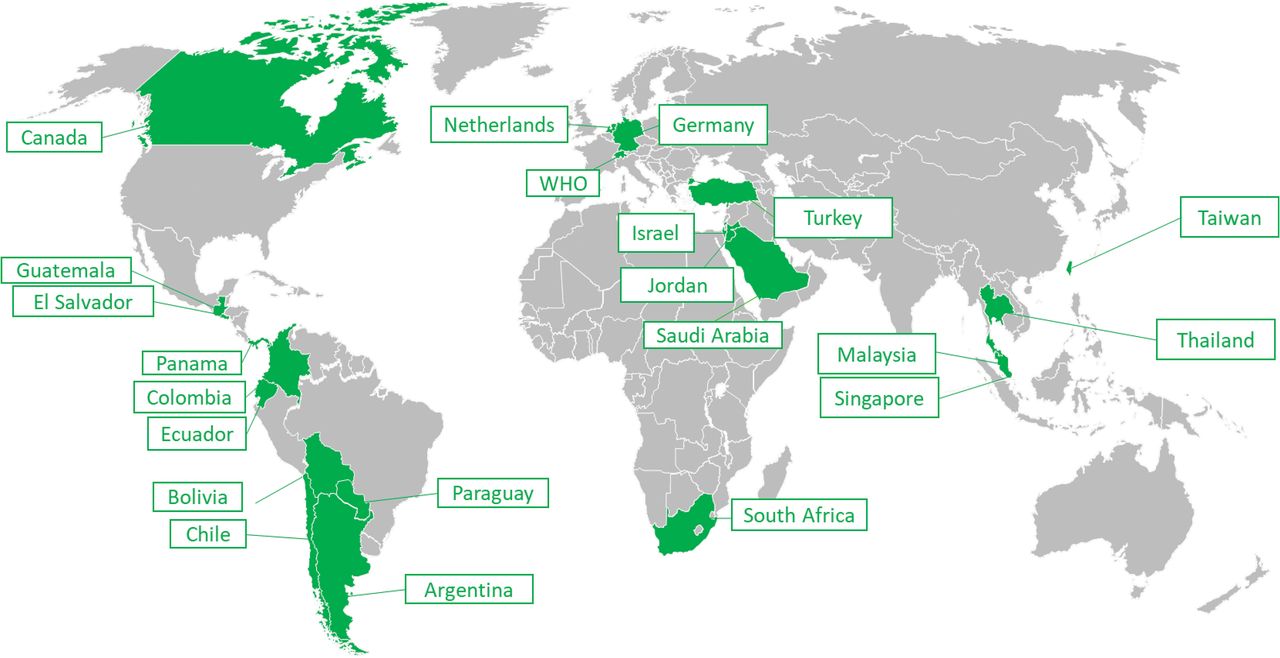
Out of the 37 NRAs in scope of the pilot, 21 (57%) accepted to participate while 11 declined and 5 neither accepted nor refused. Figure 2 illustrates the distribution of NRAs across the world who accepted to participate in the pilot.

List of NRAs across the world that participated in the pilot.
At the outset, the company did not expect all countries to participate, and one of the objectives of the pilot was to uncover barriers to the reliance principles, such as regulation or legislative challenges, which would need to be addressed for it to be successful moving forward.
Countries, such as the Dominican Republic, Peru, and Georgia classified this change as a new marketing authorization (as only one manufacturing or batch release site is allowed within one license). As a result of this classification, they were not able to participate in the pilot. India, Vietnam, and the Philippines assessed that their current or evolving regulations did not allow them to participate in this pilot at this time. Agency reorganizations in Panama and Uruguay precluded participation. Hong Kong and Indonesia preferred to use their already existing reliance pathways. Brazil was not able to participate in the pilot at this time. As the Brazilian health regulatory agency (ANVISA) explained at the Conference of the Pan American Network for Drug Regulatory Harmonization (PANDRH) held in November 2022 (27), ensuring equity between multinational companies and local companies in the way priority is given for evaluating dossiers requires that all submissions are assessed in a chronological order. Thus, participating in the pilot would have equated to bypassing the queue. The five remaining eligible countries (Azerbaijan, Costa Rica, Honduras, Mexico, and Venezuela) did not respond in the allotted timeframe. These insights provided valuable information on barriers to be addressed to enable reliance principles moving forward.
Adjustments
Some participating NRAs expressed specific requests to the company to participate in the pilot. One of the main conditions for the pilot to take place was to standardize the submission processes as much as possible (“one size fits all”). Thus, specific requests from each NRAs were carefully reviewed by the company to assess whether some minor adjustments could be acceptable or not. Table I indicates what those requests were and the company’s position for each of them. Other NRAs requested information that was considered as already included in the dossier, not applicable or without any impact on the submission process.
Specific Local Requests from Participating NRAs and the Company’s Position
Discussion
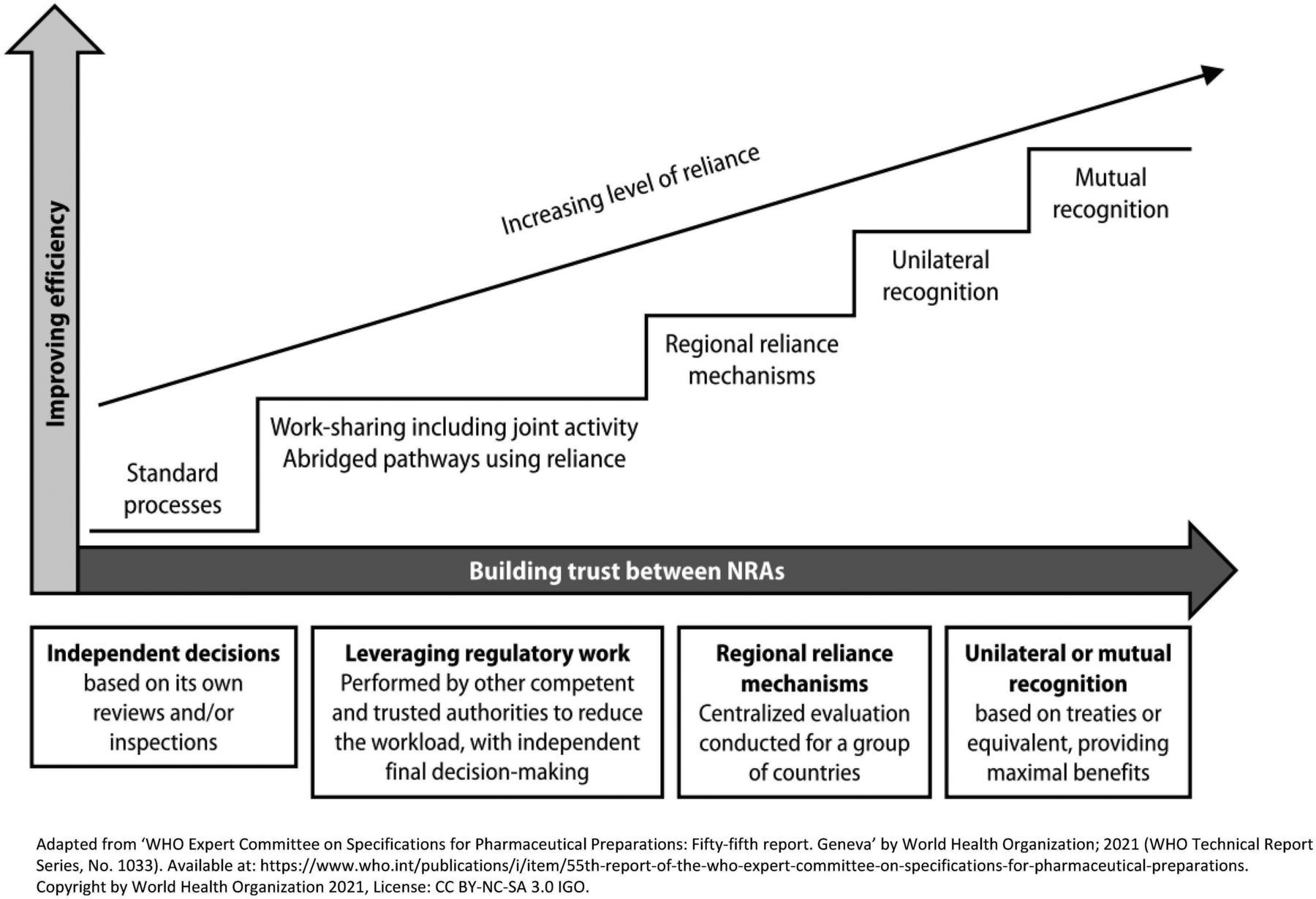
Over the last decade and even more so in recent years, the number of requirements for CMC PACs has increased due to the development of NRAs regulatory capabilities. However, the overall duration of the review and approval processes has not significantly improved. This triggers an increased risk of supply disruptions or discontinuations of medical products, and in particular, vaccines. The reliance mechanism is a solution that allows the development and upskilling of NRAs and the simplification of CMC PACs management for pharmaceutical companies and NRAs. In addition, it allows for a faster implementation of changes, which leads to a decreased risk of shortages, while allowing NRAs to maintain sovereignty in the final decision. The relying agency retains the final decision of whether to approve the variation or not. Reliance could be a first step before the development of “recognition” mechanisms as depicted in the GRelP (Figure 3).

The key concepts of reliance as per WHO guideline on good reliance practices.
The reliance mechanism is considered by WHO as one of the main factors in significantly improving the management of CMC PACs by helping global convergence and harmonization and making the best use of resources, both at the regulators and industry levels (26). Sanofi’s decision to launch a worldwide pilot for reliance using a real case study of a PAC for a vaccine product was very timely.
The lessons learned from Step 1 of the pilot, which consisted of engaging the reference authority and the NRAs, are summarized following:
At the Company level:
○ As affiliates engagement is sometimes challenging, proposing a new framework to NRAs instead of the strict application of existing legislation can work
○ Close coordination with all affiliates helps develop and provide material for the NRAs, ensure the consistency of the message delivered to each of them, and convince them to join the pilot
○ Precise timeline coordination with all affiliates is essential to move toward a specific submission date
○ Agility and creativity are required to develop an ad hoc “proof-of-concept” tool and the associated training for the purpose of this pilot
Great support provided by the four external key stakeholders (Health Canada, WHO, the German PEI, and PAHO), especially their willingness to participate and to engage a maximum of NRAs and to review and support the scientific justification to standardize the dossier
Flexibility and pragmatism of Health Canada as the reference organization:
○ Agreed to adjust the dossier requirements without compromising regulatory evaluation
○ Reviewed and approved the dossier 1 month ahead of the Canadian timelines
○ Provided the unredacted assessment report within 1 day after the Company asked for it
○ Provided the Certificate of Pharmaceutical Product (CPP) within 19 working days instead of 28 working days. However, to obtain the legalization of the CPP, a requirement for many countries participating in the pilot, 2–3 months is required. The company has committed to providing the legalized CPP during the review as soon as it is available. Considering the additional and significant time it takes for manufacturers to obtain a legalized CPP, and the fact that the NRAs in scope of this pilot are performing their own review and approval, we question the utility of providing a CPP while the intended objective of the CPP is to provide sufficient confidence to NRAs that the product they import is of the appropriate quality, safety, and efficacy in the absence of a full review, which is not occurring
Overall, 57% of the NRAs impacted by this CMC PAC agreed to participate in the pilot, illustrating the interest of numerous health authorities across four continents to test ways of improving the management of PACs. This provides incentive to continue advocating for the broader use of reliance principles. Out of the 11 NRAs who declined, 4 will either apply the reliance principles in their current review and approval practices or were in agreement with the initiative, even if they were unable to participate due to ongoing changes in their regulations. For the countries that declined to participate, the main reason was the noncompliance of the approach with their local regulations.
Five NRAs did not inform the company of their decisions despite, for some of them, their adhesion to international institutions that encourage the application of reliance principles and collaborations among NRAs (e.g., ICMRA).
Conclusions
This pilot aims to demonstrate that efficiencies can be achieved using the reliance principles as well as trust can be built between not only the industry and each NRA but between NRAs as questions and assessment reports are shared across multiple Agencies facilitating more reliance opportunities.
As a concluding remark, we invite all NRAs around the world to consider adjusting their local regulations by fully endorsing the reliance principles as stated in the WHO guidelines. This would represent a significant contribution toward global convergence and harmonization which, in turn, would benefit populations by improving access to medical products, including vaccines.
Step 2 of the pilot was launched in January 2023 and will last 6 months (see Figure 1). This step represents the review period for participating NRAs. The company will also share lessons learned from this step and make proposals moving forward so that this experience can identify opportunities and barriers—on both the regulators’ and the industry’s side—on the scale up of reliance principles for the management of CMC PACs.
Conflict of Interest Declaration
All co-authors are Sanofi employees and may hold shares and/or stock options in the company. This work was funded by Sanofi.
Acknowledgments
We acknowledge Health Canada, WHO, PAHO, and the German PEI for their support in advocating for international regulatory reliance processes. Editorial assistance was provided by Manojkumar Patel and Jean-Sébastien Bolduc from Sanofi.
- © PDA, Inc. 2024




{kind=link}
{kind=link}
{kind=link}