
Issue
EU Annex I, paragraph 113 states: “The integrity of the sterilised filter should be verified before use and should be confirmed immediately after use by an appropriate method such as bubble point, diffusive flow or pressure hold.” The paragraph wording, stating a recommendation for an activity, does not allow for applicability of the pre-use/post-sterilization integrity test based on risk evaluation.
PDA's Position
Damage to an integral filter during moist heat sterilization is most commonly caused by exceeding manufacturer's recommended pressure differential and temperature parameters. This damage, if it occurs, is prominent enough that it would assuredly be detected during the post-use integrity test. Sterilizing-grade filter validation data demonstrates that pore enlargement after moist heat exposure does not occur. For sterilization via gamma radiation, the filter is not exposed to pressure differential or high temperature and filter validation data again demonstrates that pore enlargement after radiation also does not occur. In these instances, the risk of not performing a pre-use/post-sterilization integrity test is a business or loss-of-product risk and not a product quality risk. A failed post-use integrity test indicates the product should be rejected.
Pre-use integrity testing of a sterilized filter would mean manipulation of the sterilized filtrate side. Such manipulation represents a process risk elevation. Both wetting and venting manipulations are undesirable after the filter and filtrate side have been sterilized, as the integrity test itself must be performed at atmospheric conditions of pressure and room temperature, creating the potential for microbial ingress. The impact of a sterile filtrate side manipulation may lead to a breach in sterility of the system, thus adding an unnecessary residual risk to product quality and patient safety.
PDA's Recommendation
The need of a pre-use integrity test of a sterilized filter should be left to the discretion of the filter user and should not be mandatory.
The decision to perform or not perform a pre-use/post-sterilization integrity test should be made by the filter user upon thorough, documented risk-based analysis in accordance with ICH guidelines. Based on the risk analysis, a control strategy should be implemented that includes validation, in-process monitoring and control of temperatures and pressures during sterilization to ensure that the vendor recommended parameters have not been exceeded. Careful consideration and precautions must be taken to avoid the potential for microbial ingress should the user perform a pre-use integrity test of the sterilized filter.
Additional Reading
-
EudraLex Volume 4, EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use, Annex 1, Manufacture of Sterile Medicinal Products, Brussels, 2008
-
PDA Technical Report 26, Liquid Sterilizing Filtration, Parenteral Drug Association, Bethesda, MD, 2008
-
Food and Drug Administration (FDA), Guideline on Sterile Drug Products Produced by Aseptic Processing, Division of Manufacturing and Product Quality, Office of Compliance, Center for Drugs and Biologics, Rockville, MD, 2004
Appendix: Risk Assessment (PQRI Post Approval Changes for Sterile Products Working Group, 2007)
Risk is calculated as:
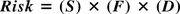
(S): Severity of the event (consequence)
(F): Frequency estimation (likelihood of event occurring)
(D): Level of detectability
The three categories are classified as:
The scenario of the pre-use/post-sterilization integrity test performed and the lack thereof are now compared side-by-side:
The lack of detection of a potential breach or an ingress or mistake made elevates the risk level when performing the pre-use/post-sterilization test.
- © PDA, Inc. 2012



