
Introduction
The purpose of this paper is to provide guidance and drive consistency in regards to microbial control for manufacturers of low-bioburden bulk biologics. This paper provides recommendations based on biologics produced using cell cultures such as monoclonal antibody (mAb)-based products and recombinant protein manufacturing process. These recommendations, from the members of the BioPhorum Operations Group (BPOG) Bioburden Working Group, are intended to assist biopharmaceutical manufacturers develop microbial monitoring strategies and product safety assessments. Each manufacturer is unique, therefore, alternative strategies may be justified and/or qualified.
Scope
This paper focuses on the following topics:
Microbial in-process monitoring during inoculum expansion
Culture expansion, and protein purification process of bulk drug substances
Setting alert/action levels limits
Objectionable organisms in bulk biologics, responding to bioburden excursions
Assessing impact to product quality
Background
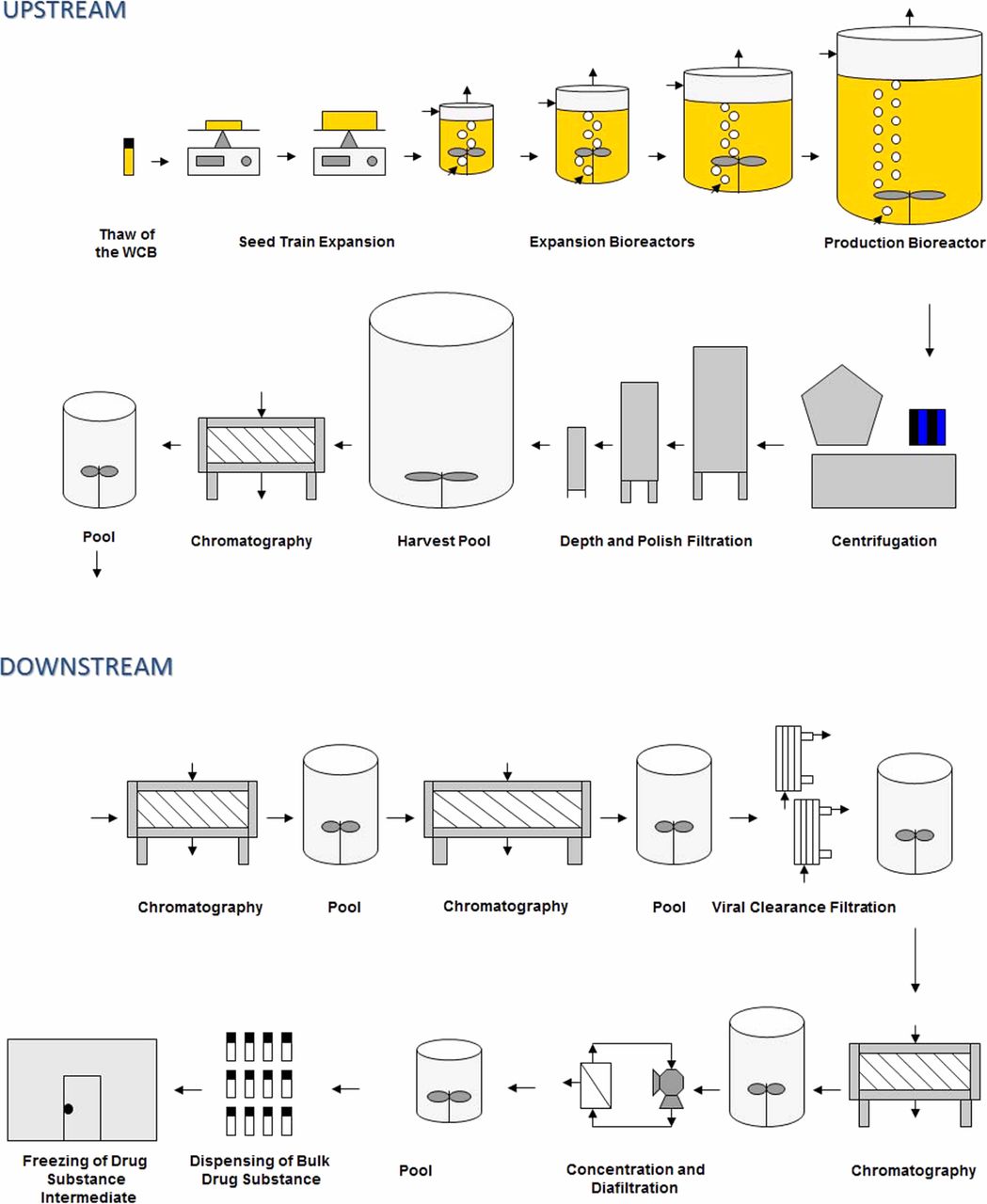
The biopharmaceutical industry produces non-sterile bulk biologics (i.e., drug substances) using bioburden-controlled processes in accordance to Q7A and Annex 2. Sterile final dosage forms are produced in accordance to Annex 1. A mammalian cell mAb process consists of upstream and downstream processes. Upstream operations include the protein production phase of manufacturing where the host cells are grown to generate the product molecule. Primary recovery (centrifugation and depth filtration) is the first step in removing the unwanted production components while retaining the product molecule. Capture of the target molecule is often achieved with affinity chromatography. Some firms chose to perform capture as a part of downstream manufacturing. Downstream operations, which typically include chromatography, viral clearance, concentration, and diafiltration, progressively refine the product to its final bulk form suitable for manufacturing into the final drug product.
The diagram in Figure 1 illustrates a generic mammalian cell mAb process.

A generic mammalian cell mAb process.
In-process Product Stream Monitoring and Testing
A comprehensive in-process sampling and testing plan is necessary to monitor and control the biological manufacturing processes. The following sampling approach is typically incorporated as part of the general sampling plan. Additional sampling locations may be necessary in some cases due to specific process considerations. In general, samples for bioburden and/or endotoxin should be taken at critical process steps (e.g., pre-filtration samples) or after hold times, where bioburden could get introduced into the process or proliferation could occur. Such sampling points for bioburden and/or endotoxin should be determined and documented by performing a risk analysis of the manufacturing process (prior to establishing the sampling and testing program).
Sample Collection
Bioburden/endotoxin sample containers and procedures are key elements of the overall monitoring program given the potential risk of false-positive and false-negative results. Materials must be well mixed just prior to sampling.
Containers used for bioburden sampling must be sterile. The recommended containers include disposable systems that are closed to the environment and/or sampling assemblies with equipment interfaces that can be cleaned or steamed with the manufacturing equipment.
Containers used for endotoxin sampling must be sterile, pyrogen-free, and made of a material that does not interfere with the recovery of endotoxin (e.g., glass, polystyrene).
Containers used for both bioburden and endotoxin sampling should be dedicated to minimize the number of times the sample is manipulated prior to testing. Where relevant, sampling flow paths may need to be flushed to ensure the process and fluids from previous sampling operations are expelled so process/sample integrity are not compromised. Technicians that perform sampling operations must be trained in proper sampling techniques. The manipulation of bioburden samples on the manufacturing floor should be minimized as much as possible prior to delivery to the quality control (QC) group, which should occur as soon as possible. Bioburden samples are recommended to be stored at 2–8 °C and tested within 24 h of collection.
Process Media Preparation
To evaluate bioburden levels associated with media preparation, it is recommended that an appropriate number of batches at scale, based on statistical analysis and/or risk assessment, are sampled/tested for bioburden/endotoxin just prior to filtration or sterilization of the media. Preferably, batches should be sampled/tested prior to process validation batches, for bioreactor media (for all bioreactors in the process). This data may be used to support the maximum hold time of media prior to sterilization. Media preparation for bioreactor steps is typically the longest preparation for upstream solutions. Where processes include complex feeds with several preparation steps (e.g., protein hydrolysates), the feeds should also be considered for sampling and testing.
Routine monitoring of bioburden/endotoxin at this step may not be needed when maximum media preparation and filtration times are established. However, periodic monitoring is recommended annually or after prolonged shutdowns.
Buffer Preparation
For filtered buffers, it is recommended that bioburden and endotoxin testing be performed on an appropriate number of batches at scale, based on statistical analysis and/or risk assessment. For final diafiltration/formulation buffers, endotoxin testing is recommended to be performed prior to use to assess the buffer's microbial quality for all batches.
All buffers which are not filtered (e.g., non-filterable buffers) should be freshly prepared and used as soon as possible. Bioburden and endotoxin sampling/testing for non-filtered buffers is recommended to be performed for all batches prior to use (buffers used for cleaning/sanitization steps may be excluded), unless a risk-based assessment justifies another approach (e.g., buffers prepared immediately prior to use, or buffers that are not growth promoting). For buffers that are stored for longer than 24 h, a hold time validation (including bioburden and endotoxin data) are expected.
Inoculum Expansion Operations
Prior to transferring from passive control (i.e., passive control of pH and dissolved oxygen) inoculum expansion to the first active control expansion step, at full scale, a bioburden sample may be taken. This sample is in addition to routine visual examination for viability and absence of microorganisms or purity check (for microbial processes). Given potential contamination risks and sample volume concerns associated with sampling at earlier inoculum expansion stages (Erlenmeyer flasks, roller bottles, etc.), sampling for bioburden is not recommended. Endotoxin testing is not routinely performed at inoculum expansion stages.
Testing for bioburden of the final culture from passive control inoculum expansion to the first active control expansion is recommended on an appropriate number of batches at scale based on statistical analysis and/or risk assessment.
Note: To support potential contamination investigations, some companies collect and retain samples at the end of each expansion operation until batch release.
Bioreactor (Inoculum and Production) Operations
Prior to transfer into the next bioreactor, a bioburden sample may be tested or kept as a back-up sample (perform a purity check in the case of microbial processes). For inoculum bioreactors, consider testing the pre-transfer culture for bioburden on an appropriate number of batches at scale based on statistical analysis and/or risk assessment. In the case of production bioreactors, all batches should be tested for bioburden at the end of production (unprocessed bulk sample). Endotoxin testing is not routinely performed at inoculum expansion stages because these stages are expected to be essentially sterile.
For continuous cell lines, bioburden samples should be taken in order to reflect the individual manufactured batches.
Harvest Operations
Clarified harvest pool (filtered or unfiltered) at the end of harvest operations should be tested for bioburden/endotoxin for all batches just prior to the start of the following process step. Only in the case when clarified harvest is filtered into a pooling tank is it recommended to test the clarified harvest prior to filtration for bioburden for an appropriate number of batches at scale based on statistical analysis and/or risk assessment. This provides adequate evaluation of bioburden control for the harvest operation (sampling pre-filtration should be performed as close as possible to the end of the harvest operation).
Chromatography and Ultrafiltration/Diafiltration (UF/DF) Operations
To evaluate column and UF/DF performance during operations it is recommended that bioburden and endotoxin testing be performed on an appropriate number of batches at scale, based on statistical analysis and/or risk assessment at the following steps:
Water for injection (WFI) rinse (at retentate or drain line) following removal of storage solution (if system is stored wet). This is a valuable sample points for evaluating column and resin storage effectiveness.
WFI rinse following pre-use sanitization step (prior to the start of equilibration phase). This is a valuable sample points for evaluating column and resin cleaning effectiveness.
Equilibration buffer at the end of membranes equilibration step (prior to the start of product concentration phase).
Note: Some events have shown that microorganisms can be bound to the resin during equilibration phase and elute with wash/elution buffers).
WFI rinse following post-use cleaning/sanitization step (prior to start of storage phase).
Each protein pool (filtered or unfiltered) at the end of chromatography and UF/DF operations should be tested for bioburden and endotoxin. When the protein pool is filtered into a pooling tank, the protein elution is recommended to be tested for bioburden prior to filtration. This ensures adequate evaluation of microbial control of the chromatography and UF/DF operations (sampling pre-filtration should be performed as close as possible to the end of the final elution step).
Microbial control of reusable filters, resin storage conditions of new and unused resins, and resin stored in columns should be demonstrated and validated. Resin storage conditions should be tested for bioburden to demonstrate bacteriostatic conditions. Acceptance criteria for these samples should be set using the vendor's specifications for new and unused resins (typically ≤100 CFU/mL).
Bulk Dispensing Operations
Bioburden and endotoxin samples should be taken from the final bulk drug substance (post-filtration). There are many different configurations for bulk filling operations. The bioburden/endotoxin samples collected should be representative of the operation, taking into consideration the risk associated with sampling and the nature of the filling operation (e.g., closed vs open filling; single container vs multiple containers).
Microbial Test Methods
Endotoxin testing can be performed using any of the approved compendial methods, although the kinetic chromogenic/turbidimetric techniques are recommended because these methods are more precise and data interpretation is automated.
Bioburden testing can be performed using any of the approved compendial methods or validated rapid microbial methods using at least 10 mL samples and should be conducted in a suitably controlled environment to minimize the potential for laboratory-introduced contamination. Given the matrix characteristics of bioreactor samples, it may be necessary to use more than one filter to process a 10 mL sample (e.g., five filters with a 2 mL sample on each filter). Use of validated rapid microbial methods is recommended to reduce time to results, which enables quicker responses to microbial excursions.
Note: Suitability testing should be performed using cultures from a recognized culture collection (e.g., ATTC) and environmental microorganisms for each sample matrix or family of sample matrixes to ensure the ability to recover low levels (<100 CFU) of microorganisms.
Additionally, laboratory managers need to ensure that bench microbiologists are adequately trained to differentiate between bacteria, yeast, and molds, as well as the presences of spreaders/confluent growth of microbes. Microbiologists should also be able to distinguish macroscopic colonial types by morphology (with or without magnification). If there is a frequent recovery of spreaders/confluent colonies, the laboratory may need to read the samples on a daily basis to determine an accurate estimation of the bioburden level.
Plate count recoveries exceeding 250 CFU on the most dilute sample are reported as too numerous to count (TNTC). A TNTC result for any in-process bioburden sample should automatically result in an action level or an out of specification (OOS), which requires an investigation. A TNTC recovery on an in-process bioburden sample should be a rare occurrence due to measures taken to prevent/minimize bioburden contaminations from occurring in biologically manufactured products. A valid TNTC result indicates that there has been a potential breach to the manufacturing controls put in place to prevent contamination from occurring. Investigations initiated in response to TNTC results need to be robust and thorough to ensure patient safety.
In the case of microbial processes, the testing used at the end of the production bioreactor is typically a purity test to confirm absence of non-host microorganisms by plating on selective and non-selective media and visual examination. The microbial purity method should also be validated using spike recovery studies. The spike recovery studies should be performed using samples containing host cells at a viable titer that is representative of the process.
Setting Bioburden Control Levels (Alert/Action)
Unlike non-sterile dosage forms, there are no recommended bioburden levels provided in regulatory guidelines or compendia for the protein purification processes of biologic or other biopharmaceutical products, therefore, manufacturers are responsible for setting bioburden control levels for biologic production processes.
The BioPhorum Operations Group (BPOG) Bioburden Working Group conducted a member survey of bioburden action levels and found that the majority of biologic processes action levels were set between 1 and 10 CFU/mL. When control levels (i.e., action and/or alert levels) are set appropriately, drifts or deviations from normal operating conditions can be promptly detected, investigated, and remediated. When control levels are not set appropriately (e.g., too loose or too tight), a loss of microbial control may go undetected or unnecessary investigations may be performed. Therefore, setting appropriate control levels is a key component of a successful microbial control strategy.
Because control levels are set prior to product licensure, they are often set with limited data, or they are based on comparable processes until enough data reflecting process capabilities can be attained. After commercialization additional data will be available, and periodic review is required to ensure control levels reflect process performance over time. Consideration should be given to correlate results of bioburden testing to results of routine environmental monitoring of the manufacturing facility and equipment. Review of control levels should be proceduralized to ensure consistency across the manufacturer's product portfolio.
Objectionable Organisms
The concept of objectionable organisms is applicable to non-sterile drug products because viable microorganisms may be delivered to the patient. However, this concept is not applicable to biologics manufacturing of sterile drugs because no microorganisms are allowed in the final dosage form. Furthermore, creating a list of objectionable organisms for biologic in-process samples may actually limit the scope of investigation and keep objectionable manufacturing conditions from being assessed and addressed.
Unlike non-sterile drug manufacturing where the processing environments are often hostile to microbial growth or include the addition of preservatives, manufacturing of biologics require growth mediums (upstream cell culture steps) that enhance bacterial growth or process buffers (downstream protein purifications steps) that are often bacteriostatic. Due to these manufacturing environments, bioburden levels in biologics must be controlled to and reach near-sterile conditions in upstream process steps and ≤ 10 CFU/mL during downstream processing.
Responding To Bioburden Excursions
When bioburden control levels are exceeded, an investigation with the rigor commensurate with the risk (e.g., action level vs alert level excursion) should be conducted. To ensure adequate and consistent responses to bioburden control level excursions and adverse trends (variation from the historical mean over time), manufacturers should establish written procedures for the investigation of these events. The following are recommended actions to take in response to control level excursions:
Adverse Trends
When adverse trends are noted they should be investigated, using at minimum, a comparable approach as action level excursions (see below).
Alert Level Excursions
Alert levels are set to monitor process performance and provide early warning of an adverse trend or action level excursion. As such, the following actions are recommended:
Initiate an investigation record in the quality system.
Review historical data for adverse trend.
Identify microorganisms to species level, if possible, for microorganism trending purposes.
Notify affected departments and the quality assurance group and/or as defined by local procedures.
Action Level Excursions
Action levels are set to ensure the process is operating as designed, and they require investigation and remediation if not met. In addition to the actions required for alert level excursions, the following actions are recommended to determine the cause of the excursion, evaluate the significance of the excursion in the contexts of other data, and to initiate action to restore intended operating conditions.
Initiate an investigation record in the quality system.
Obtain feedback from a cross-functional investigational team (e.g., manufacturing, quality control, quality assurance, facilities, engineering, et. al.).
Perform concurrent laboratory and manufacturing investigations.
Determine root cause (e.g., review utilities, compressed gases and WFI systems, equipment maintenance, engineering changes, environmental monitoring, training, etc.).
Assess potential product quality impact.
Implement corrective and preventive actions (CAPAs).
Perform CAPA effectiveness check, where appropriate.
Note: Bioburden excursions that meet or exceed the action level do not necessarily indicate that product quality has been compromised, but they do indicate the need to investigate. Consider conducting a risk assessment to evaluate if the process should be halted pending resolution of the issue and completion of a return-to-service plan.
When bioburden action level excursions or adverse trends are noted, product safety and product stability need to be assessed. The following information can be used for this assessment, in addition to other factors:
What organism(s) was recovered? When possible, identify the species level.
Is the disinfectant procedure effective at removing the recovered organism, if applicable?
What toxins and/or microbial byproducts does the organism(s) produce or release?
What stage in the process did the excursion occur?
What downstream purification steps were performed after the organism(s) was recovered? Are the purification steps validated to remove bioburden? Is there data that supports clearance of the possible microbial byproducts?
Are there any connections or links to other bioburden excursions in either upstream or downstream processes?
Have the same organism(s) been observed during the production of the previous lot?
How long was product held at the step (residence time of organism) where the excursion was detected and at what temperature and at which pH?
Were changes made to the manufacturing process (e.g., equipment modifications) prior to the excursion(s)?
What potential impact could the recovered organism(s) have on the manufactured protein?
What is the impact of the microbial byproduct production (toxins) on the patient or product safety? Is there stability data that would provide assurance that the drug substance or drug product has not been affected?
Are all in-process control (IPC) results within historical trends, and does the drug substance meet all release specifications?
What information was gathered from literature searches and/or during studies conducted during the investigation?
Assessing Impact To Product Quality
Assessing residual microbial byproducts related to patient and product safety
Example 1
For adverse trends or action level excursions, the following areas should be assessed and may be included in an investigation:
Perform laboratory investigation, which should include, but is not limited to, the review of analyst's training, calculations, negative controls, sampling handing, and test materials.
Review heating, ventilation, and air conditioning (HVAC) performance in the respective manufacturing area, that is, last filter certification reports, differential pressures for date under investigation, and trend history.
Review environmental monitoring and critical utility data for the respective manufacturing area.
Review work order system to examine operation of equipment for anomalies (e.g., belt grinding and producing elevated total particulates).
Review cleaning and sanitization of the impacted area, including agents used, their preparation, and their effectiveness against the recovered organism(s).
Review any changes to the facility, utilities (e.g., compressed gases and WFI systems), equipment (e.g., chromatography skids, column packing operations, etc.) or process (e.g., logbooks and work orders).
Review batch record for any interventions that may have contributed to the excursion(s).
Conduct interviews of the personnel involved, that is, manufacturing associates, and laboratory analysts.
Confirm microbial identification to the species level. If the organism is the same as previously recovered in the process, perform strain typing to determine if it is from the same source.
Conduct a literature search on the recovered microbe to determine the worst-case toxin that might impact patient safety and publish in peer reviewed journals.
Determine if the organism strain recovered has the gene encoding the worst-case toxin.
Determine if the worst-case toxin is expressed in the specific condition in which the recovered microbe was isolated.
Additional suggestions:
Determine if the level of the worst-case toxin in subsequent processing unit operation(s) can be reduced using a scale-down model.
When there is not an identifiable worst-case toxin, use a surrogate toxin (protein) for data gathering and risk assessment.
For bulk drug bioburden excursions, perform filtration risk assessment of the product-specific validated retention capacity to calculate safety factor.
Example 2 (Model developed by Roche)
Though bioburden is easily removed by filtration steps during the purification processes, residual microbial byproducts may possibly be co-purified with the product and have detrimental effects on patient and product quality. Quality impact assessments should include the following:
Criticality of the process step
Excursions during cell culture are not acceptable, and if contamination is confirmed then the batch should be rejected.
Review of trend data
Ensure data provides assurance that there is no systemic failure or evidence of biofilm.
Assessments of microbial byproducts that can have adverse effects on patient safety. These are primarily:
Exotoxins:
Protein exotoxins (e.g., Botulinum Toxin A)
Non-protein exotoxins (e.g., Aflatoxin)
Endotoxin
Flagellin
Microbial DNA
Cell wall polysaccharides
With the exception of endotoxin, there are no available good manufacturing practice (GMP) assays to detect these microbial byproducts. However, worst-case calculations of the levels of these components can be performed and compared to a specific safety level using the following:
Identity of the contaminant(s). Identification by genotypic methods is highly recommended.
Literature search on the contaminants for cell size and known exotoxins.
The level of the contamination (CFU/mL)
Concentration of the drug substance
Maximum dosage
An example of such a calculation follows.
Scenario:
A downstream purification pool is contaminated with 100 CFU/mL of Bacillus cereus.
Assumptions:
Protein concentration of the drug substance: 22.5 mg/mL
Dosing: Product has a maximum dose of 750 mg
Conclusion
This paper provides recommendations from a biopharmaceutical industry perspective on microbiological monitoring of in-process intermediate products, including drug substances. Additionally, recommendations on subjects commonly encountered in the establishment of a monitoring program, such as setting alert/action levels, objectionable organisms in bulk biologics, responding to bioburden excursions, and assessing impact to product quality, are included.
These recommendations assist biologic manufacturers in refining their current microbial control strategy, as well as developing control strategies for new processes and products. Establishing appropriate microbial control levels provides indications of the effectiveness of the manufacturing process.
Responding to bioburden excursions can be difficult and are often inconsistent within the same company. This paper provides information that can be used to develop an investigational checklist that will help drive consistent and thorough investigations, and provides examples for assessing impact to product quality when bioburden excursions occur. To ensure consistent impact assessments and successful regulatory review of microbial excursions, formal impact assessment models are recommended.
An important aspect of all impact assessment models is the treatment of objectionable organisms. The BPOG members who contributed to this paper believe the concept of objectionable organisms is not applicable to biologics, as all organisms recovered in in-process and drug substance samples should be identified to species level, and trended and investigated thoroughly when recovered above the action level. Furthermore, final drug product formulations are sterile, which greatly reduces the risk of delivering microbes to the patient.
In the future, the BPOG Bioburden Working Group will use this paper to present an industry perspective on microbial monitoring and control of biologic manufacturing processes to regulatory agencies to gain their acceptance and to influence future regulations of non-sterile bulk biologic manufacturing.
Acknowledgements
We thank the following for their help and support: Marc Kinnelly of Astra Zeneca, Bastian Omokoko and Eileen Economy of Bayer, Barbara Daddis of Bristol-Myers Squibb, Jeri Bonilla of Genentech, Jay Stout and Beth Junker of Merck, Cheryl Mowen of Novavax, Randall Thompson and Cathleen O'Connor of Shire, Friedrich von Witzingerode of Roche, Jose A. Marrero of AbbVie and David Bain of the BioPhorum Operations Group (BPOG).
Footnotes
BPOG SPECIAL SECTION: The following article is a special editorial contribution from the BioPhorum Operations Group (BPOG). Please note that it did not go through the PDA Journal and Pharmaceutical Science and Technology regular peer review process.
- © PDA, Inc. 2015
References
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.




{kind=link}
Jump to section
- Article
- Introduction
- Scope
- Background
- In-process Product Stream Monitoring and Testing
- Inoculum Expansion Operations
- Microbial Test Methods
- Setting Bioburden Control Levels (Alert/Action)
- Objectionable Organisms
- Responding To Bioburden Excursions
- Assessing Impact To Product Quality
- Conclusion
- Acknowledgements
- Footnotes
- References
- Figures & Data
- References
- Info & Metrics
Related Articles
Cited By...
- Quantitative and Qualitative Evaluation of Microorganism Profile Identified in Bioburden Analysis in a Biopharmaceutical Facility in Brazil: Criteria for Classification and Management of Results
- Risk-Based Selection of Environmental Classifications for Biopharmaceutical Operations
- Mold Control and Detection in Biological Drug Substance Manufacturing Facilities: An Industry Perspective