
Introduction
PDA's PAC iAM (Post-Approval Change: Innovation for Availability of Medicines) program has been established with the objective to identify, assess, and address current barriers to implementation of post-approval changes (PACs) that are intended to ensure continued operations, maintain a state of control, and drive innovation and continual improvement. International Conference on Harmonisation (ICH) Q10 Annex 1 provides the basis for more effective post approval change management: “When companies can demonstrate an effective PQS [pharmaceutical quality system] and product and process understanding, including the use of QRM they “gain the opportunity to optimise science and risk based post-approval change processes to maximise benefits from innovation and continual improvement” (1).
As part of the effort to identify, assess, and address current barriers to PACs, the PDA administered a global, blinded, cross-functional survey to document industry experience with PACs in the current global regulatory environment, and to better understand the regulatory complexity and burden on the industry. The survey interrogated on specific points related to annual volume of PACs, reasons, time commitments/cycle time, impact of regional differences on change implementation, current use of tools (e.g., Post-Approval Change Management Protocols, PACMPs), impact on supply chain complexity (e.g., inventory, variants to manage, non-compliance to filings, drug shortages), manufacturing innovation, and resources needed.
Although both industry and regulators have recognized the need for improving speed and reducing complexity of PAC management to address the situation [e.g., ICH Q12, World Health Organization (WHO) efforts], only limited data—mostly in the form of isolated case studies or anecdotal subjective information—is currently available to drive the change. This survey was intended to quantitate the complexity of global PAC management, to drive awareness of the current challenges of PACs, to support ongoing efforts to ease the post-approval regulatory complexity, and to pave the way for innovation and sustainable supply of quality medicines to patients.
Summary
The 2017 PAC iAM Survey confirmed many of the perceptions and anecdotal experience within industry and regulatory authorities about the complexity of PAC management. Data from the survey confirms that many organizations deal with long change approval timelines, multiple item versions, or variants of products to manage and multiple global regulatory submissions. This complexity has the unintended consequence of creating drug shortage and supply issues, compliance issues, and more importantly resulting in organizations foregoing innovation and process improvements. The data supports the ongoing efforts from both regulators and industry to ease post-approval regulatory complexity, accelerate innovation, and ensure sustainable supply of quality medicines to patients.
It is noted that the use of PACMPs or comparability protocols in the United States (US) and European Union (EU) markets provides some level of relief from PAC complexity; however, as these approaches are not accepted globally, the overall burdensome situation remains unchanged, and the barriers to continual improvement, innovation, and technology advances remain high and insurmountable in many cases. Steps towards global convergence of regulatory requirements and leveraging additional mechanisms such as the companies' pharmaceutical quality system (PQS) for change management are needed to ensure our industry continues to move forward and provide sustainable supply of product to patients.
Data from this survey point to the urgent need to find global solutions and are in support of initiatives currently pursued at international level by ICH, the WHO, and International Federation of Pharmaceutical Manufacturer & Associations (IFPMA).
Participation
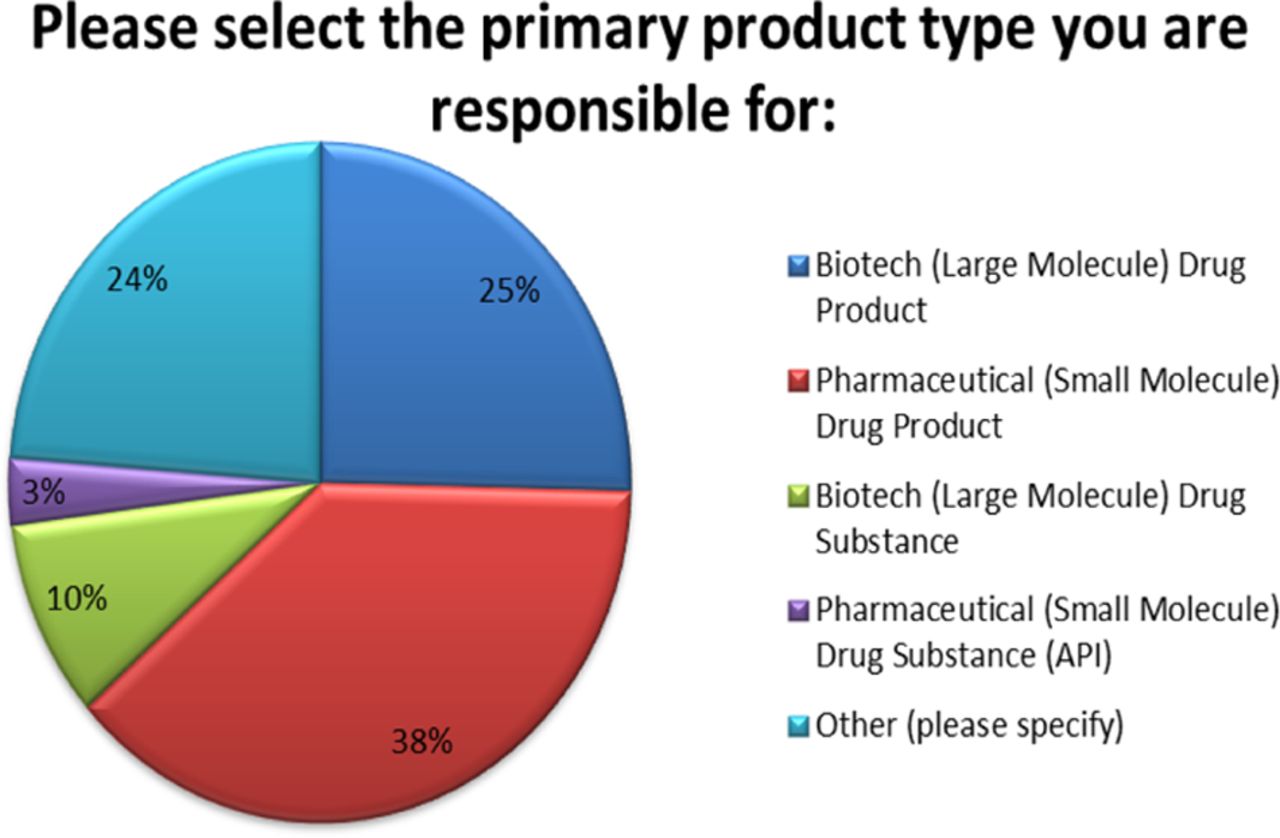
Eighty-five (85) respondents from Quality, Regulatory, Manufacturing, Technical Operations, and Development provided input on a variety of PAC-related topics including the number of PACs generated, reasons for PAC, inventory implications, and timeframes for approval. These professionals spanned from several industry sectors including Large Molecules Drug Product (DP), Large Molecule Drug Substance (DS), Small Molecules DP, and Small Molecules DS (active pharmaceutical ingredients, APIs), as well as other sectors including cell and gene therapy products, vaccines, and combinations of the above (see Figure 1).
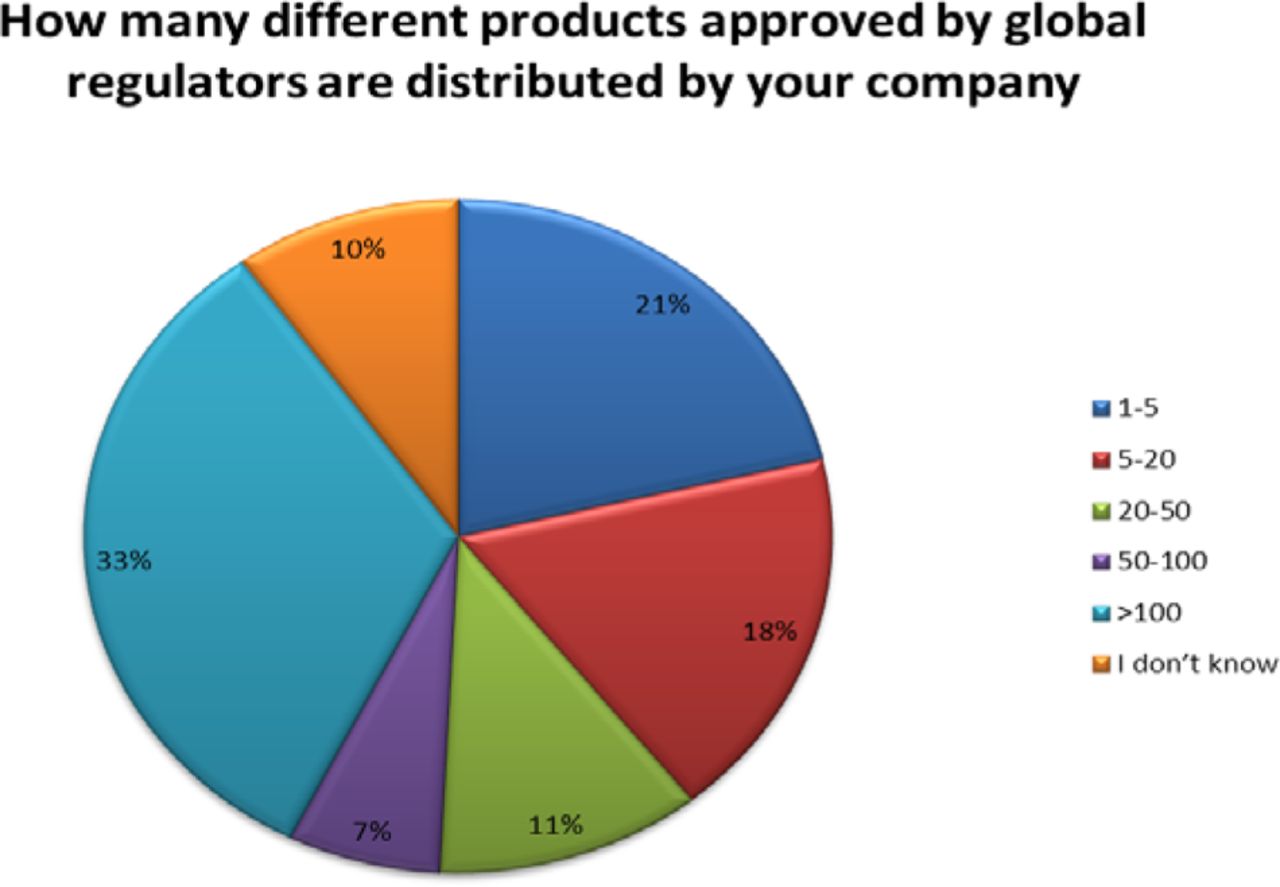
The size of these organizations varied; however, 51% of the companies responding market more than 20 products with 33% marketing over 100 products (see Figure 2).
Most PACs Reflect the Need to Manage Capacity, Innovate
A product's commercial life spans 20+ years, and increased product knowledge during this lifespan leads to better product and product understanding. In order to manage the lifecycle of a product during this long commercial lifespan, changes are inevitable to reduce risk, incorporate new product/process understanding, and drive continual improvement. The survey indicates that PACs for a product are driven by many different factors (Table I). A large percentage of organizations make changes to increase or reduce manufacturing capacity (76%), add or change manufacturing sites (73%), and comply with new regulations (53%). It is encouraging to note that, despite the complexity of the current PAC environment, many changes are being made to improve processes (89%), introduce innovative technologies (60%), and upgrade or replace obsolete equipment (71%).
Why Does Your Company Make Post Approval Changes? (Check All That Apply)
Complexity—Timelines and Product Variants Contribute to Inventory Segmentation/Drug Shortage
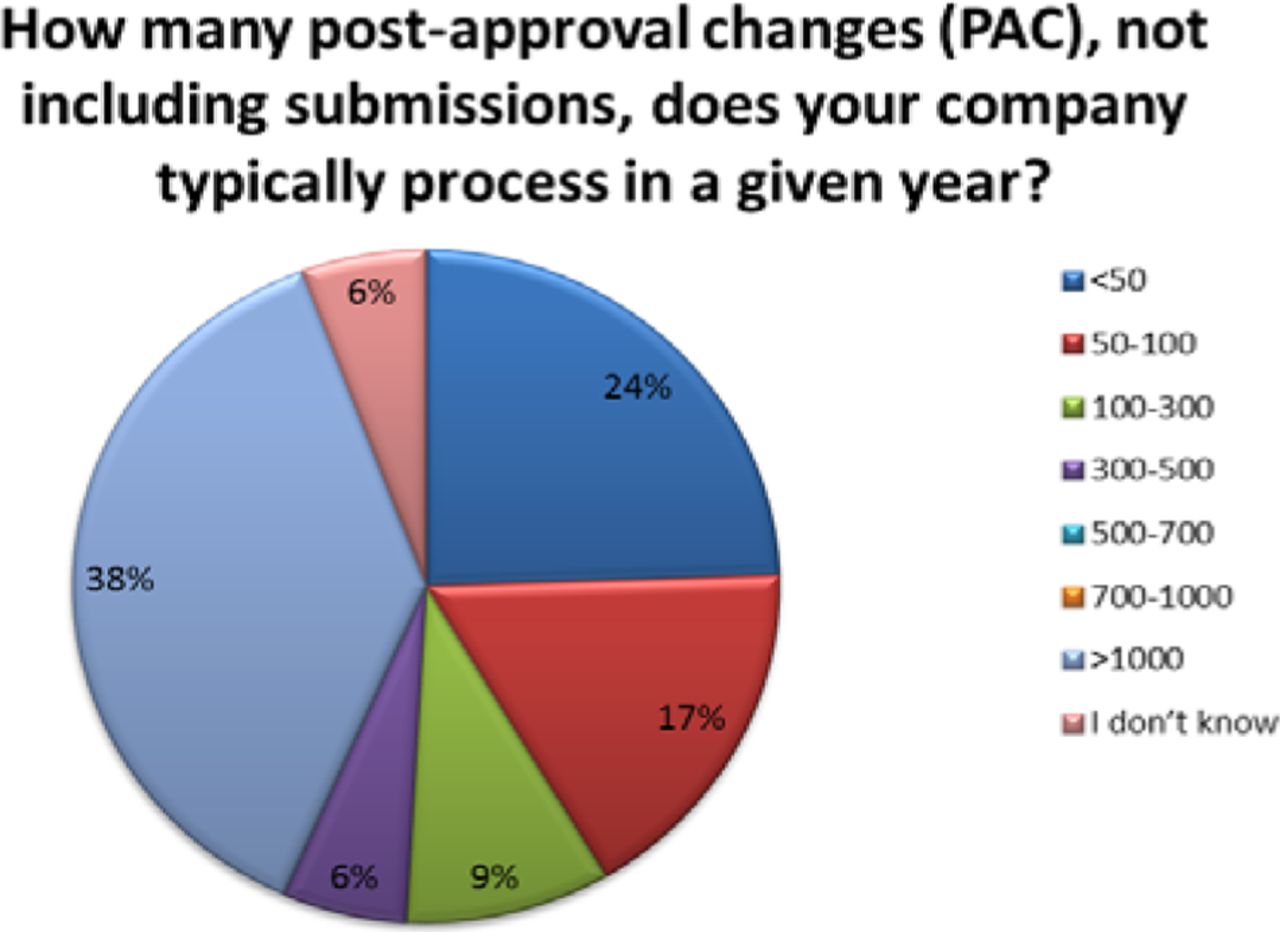
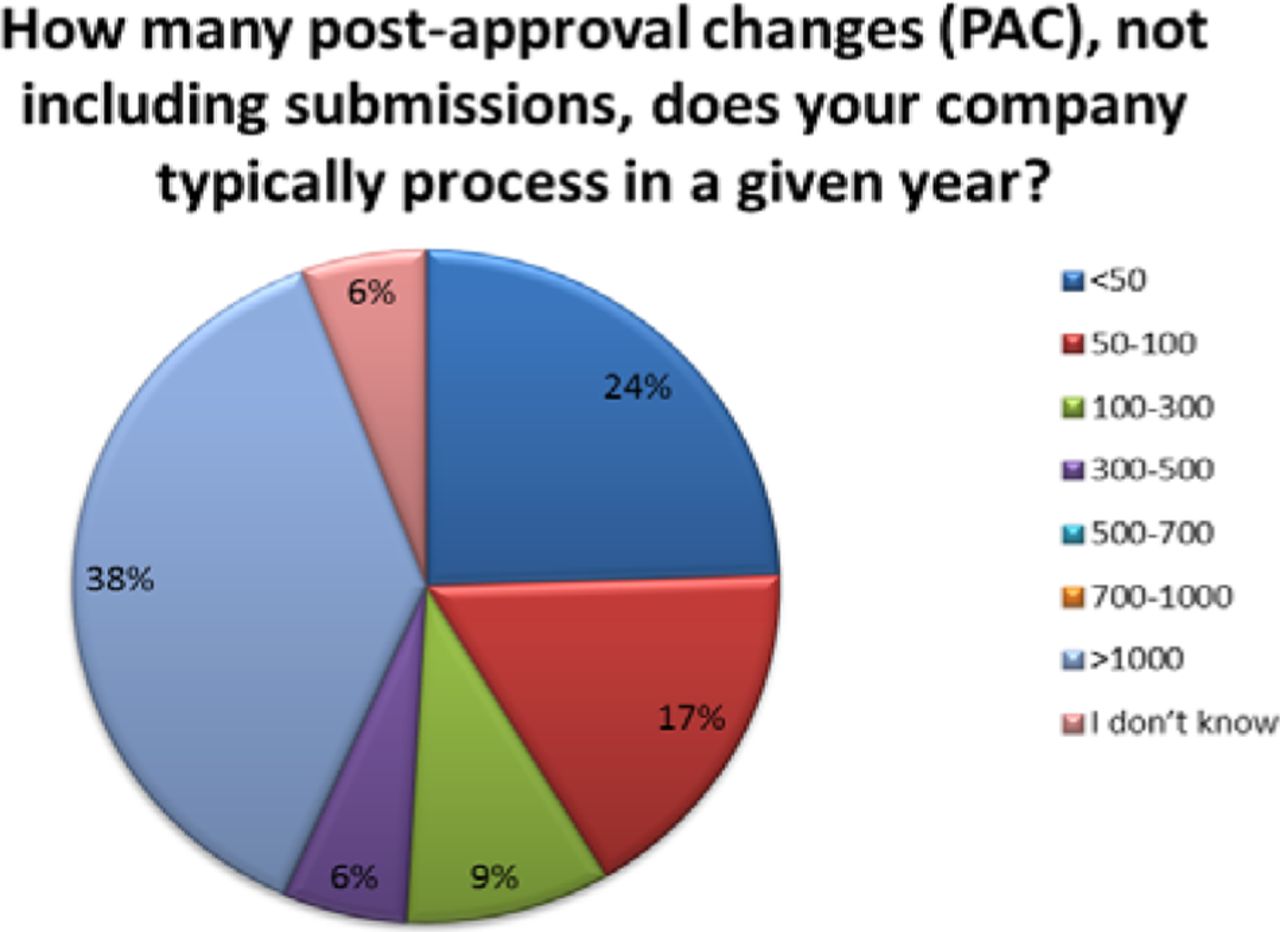
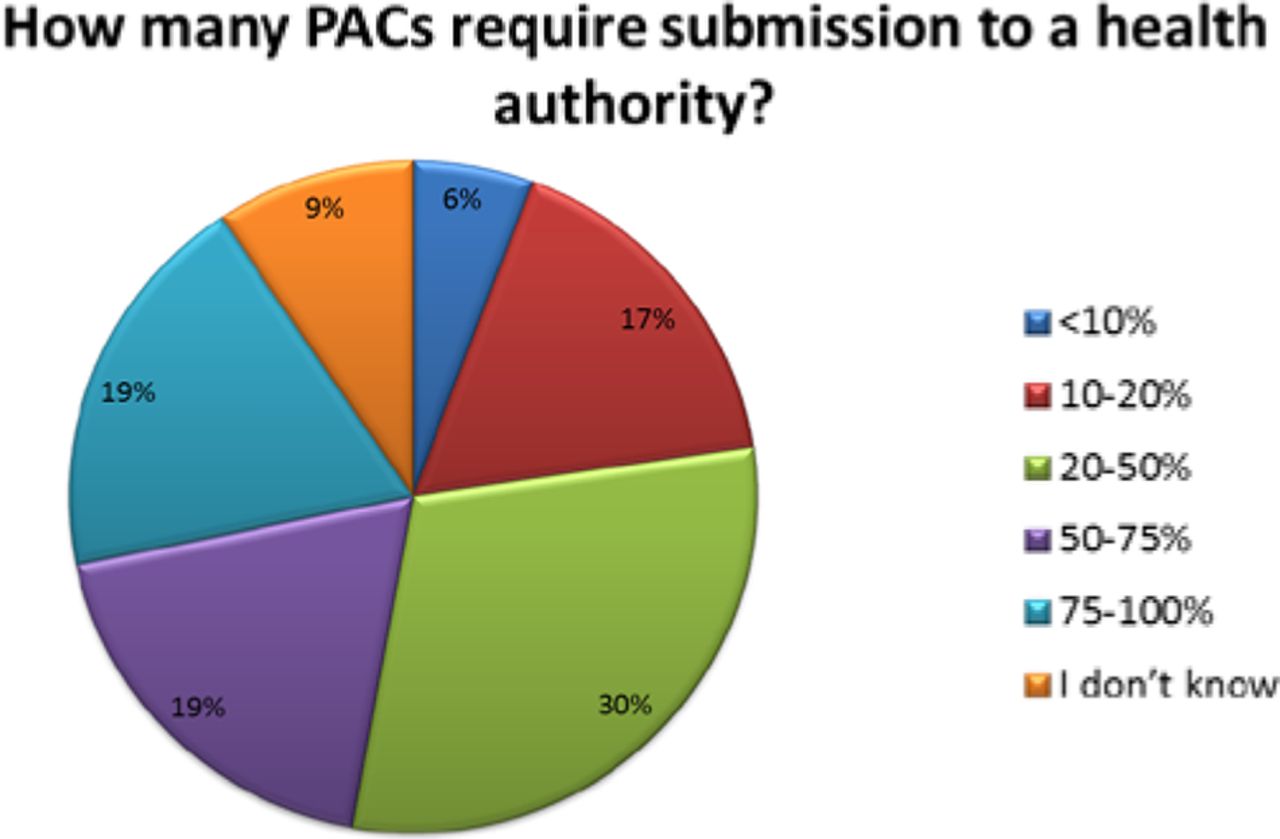
The number of PACs processed in a year varied across organizations. Thirty-eight percent (38%) of respondents reported handling over 1000 PACs in a given year, with 24% processing 50 or fewer and 32% processing between 50 and 500 changes—these are high annual volumes of PACs (see Figure 3). Importantly, almost 40% of companies indicated that greater than 50% of their changes required submission, meaning that many changes can't be managed solely within the company's PQS (see Figure 4).

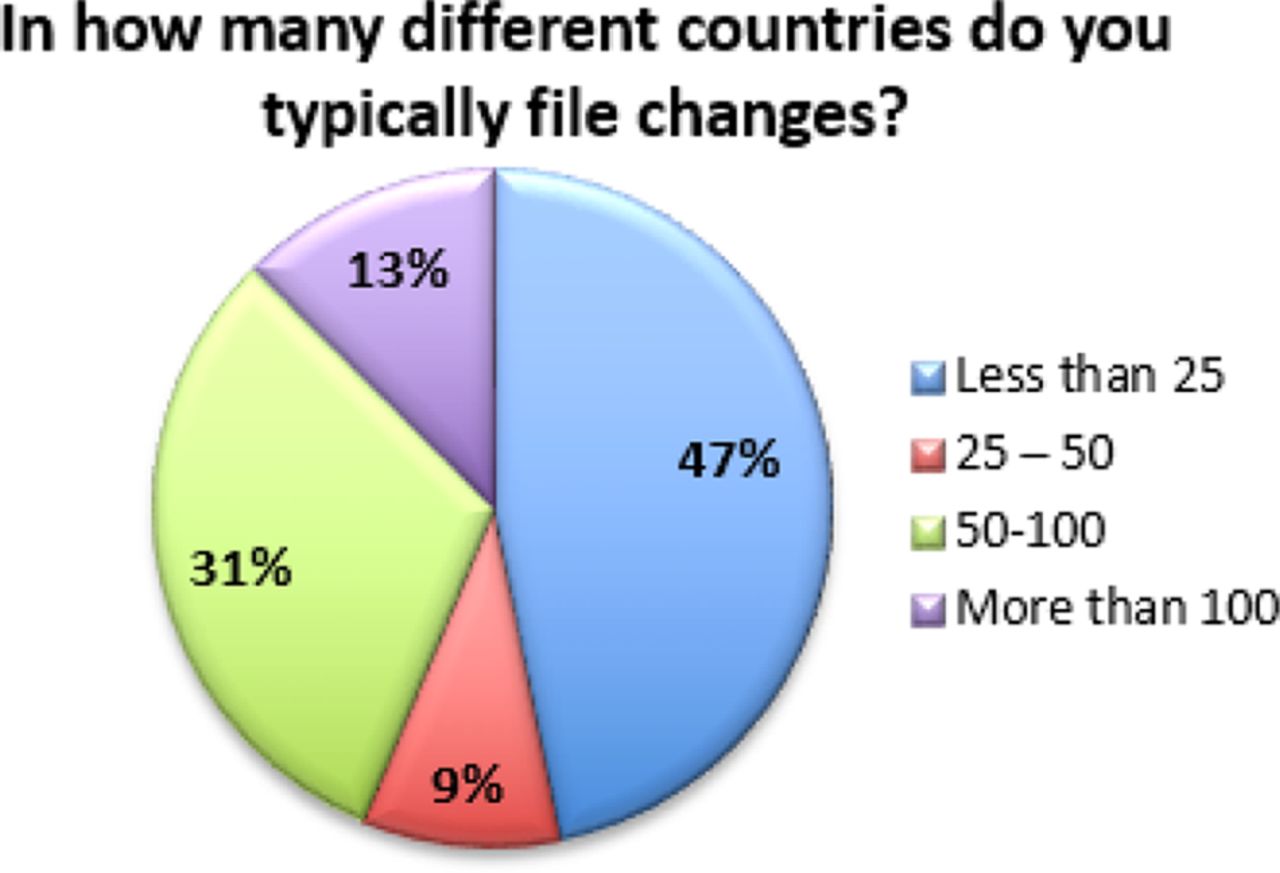
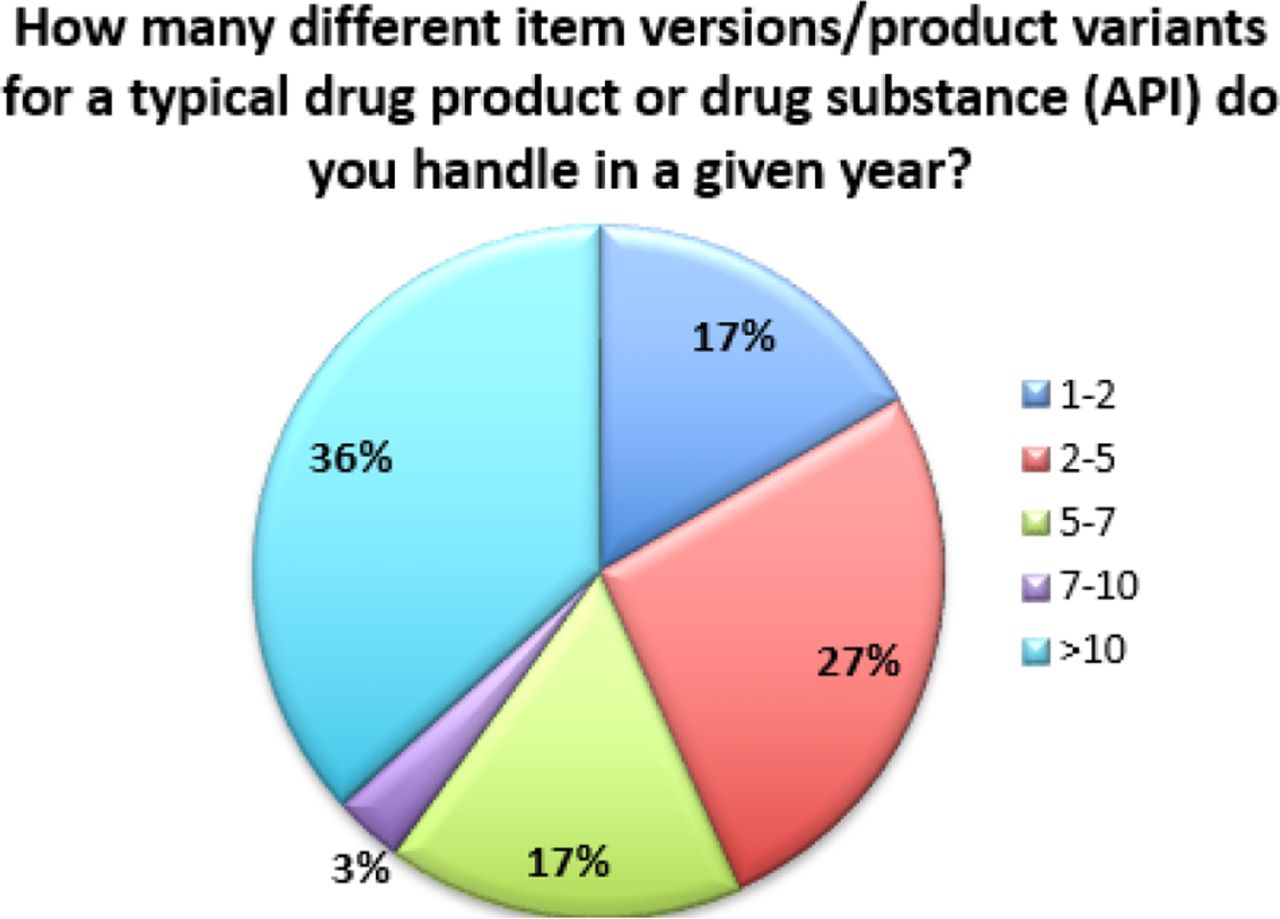
Additionally, the data indicates that many of these changes are considered moderate or major, and thus require prior approval from regulators. Fifty-seven percent (57%) of respondents indicate that over 20% of the changes processed require prior approval from regulators (see Figure 5). Combined with the fact that 53% of respondents also file changes in 25 or more countries (with 13% filing in more than 100 countries), this exponentially increases the complexity of managing a single change (see Figure 6). In fact, over 35% of respondents are managing more than 10 product variants (item versions) for each DP or DS (API) in a given year (see Figure 7). Management of multiple product variants requires additional resource to manage product release and increases the risk of mix-ups and non-conformance to regulatory dossiers.

Time Required for PACs
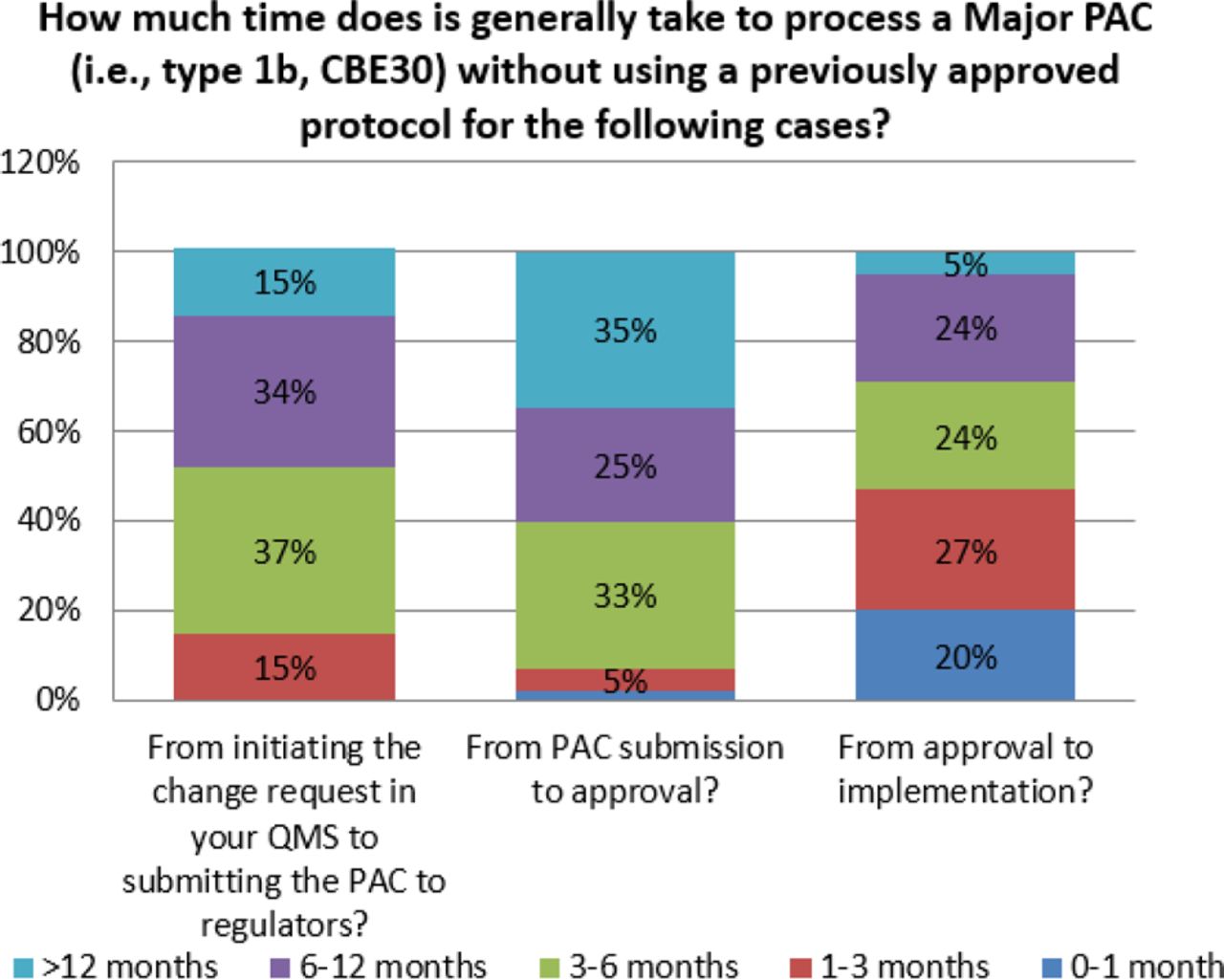
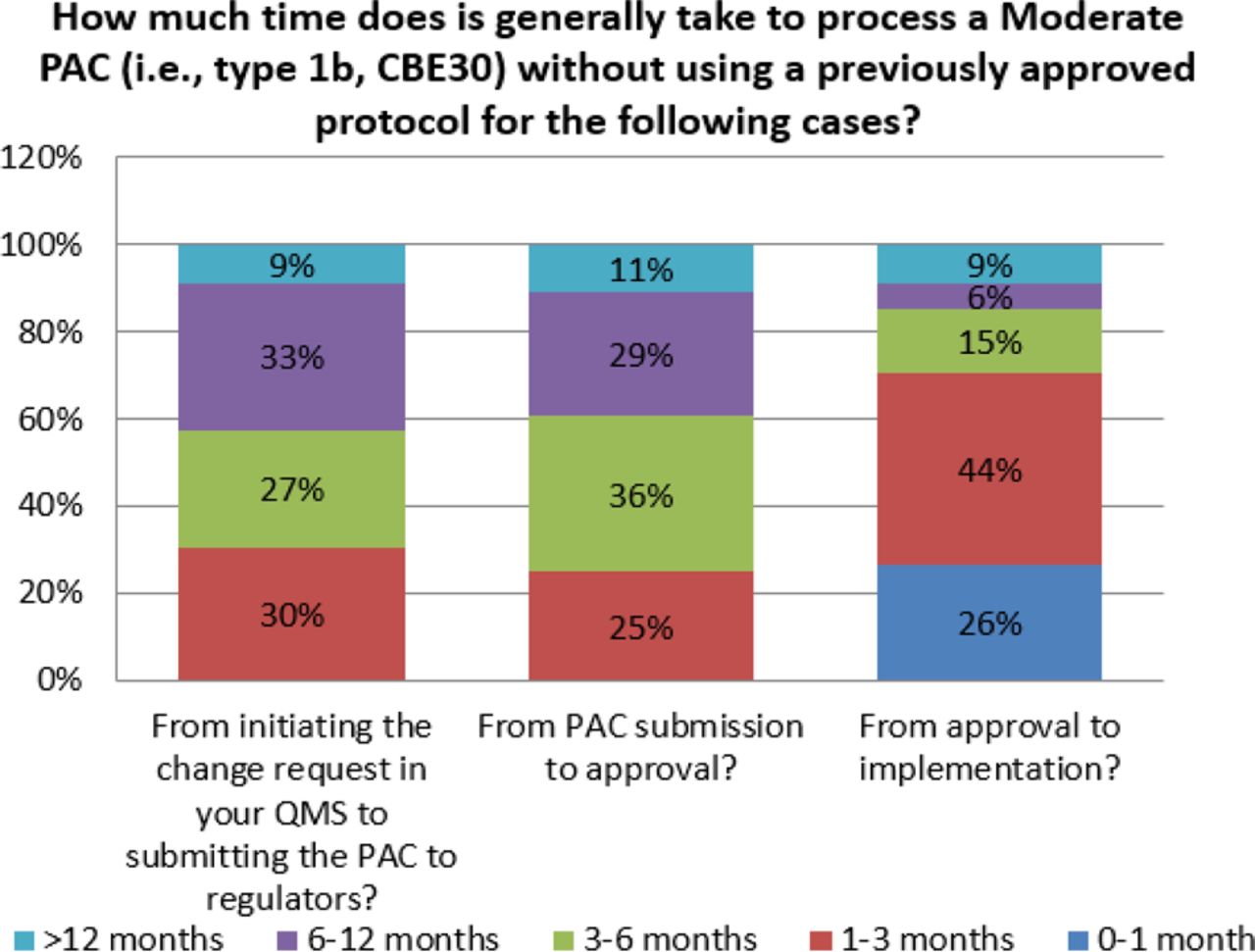
For some changes, the time from submission to approval can be excessive. Survey responses indicate that for major changes, 35% of organizations indicate that global approval can take over 12 months (see Figure 8). This is observed even for moderate changes, albeit at a lower frequency (11%) (see Figure 9). When this is combined with the number of markets, both major and moderate changes can take many years and increase the number of variants in inventory for a given product. This complexity contributes to additional inventory segregation and thus increases the potential for drug shortages, and it also creates a significant disincentive to make even high value-added changes.
Post-Approval Change Management Protocols (PACMPs) May Help Lessen the Burden
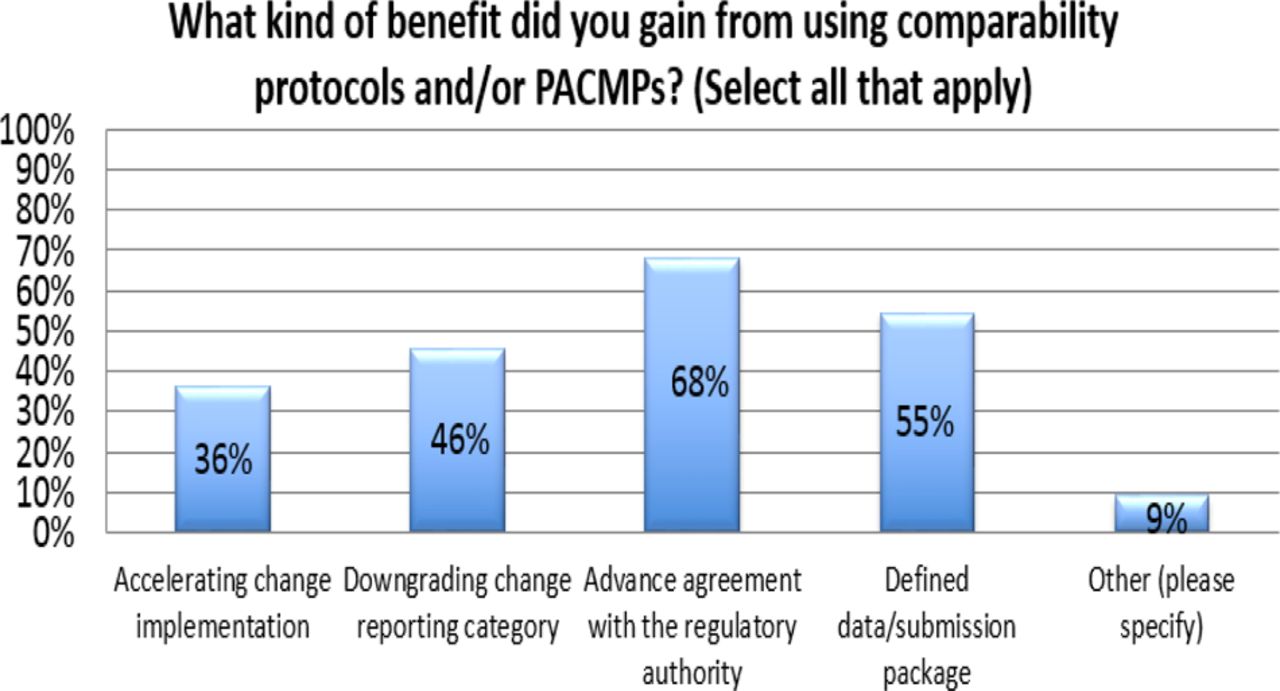
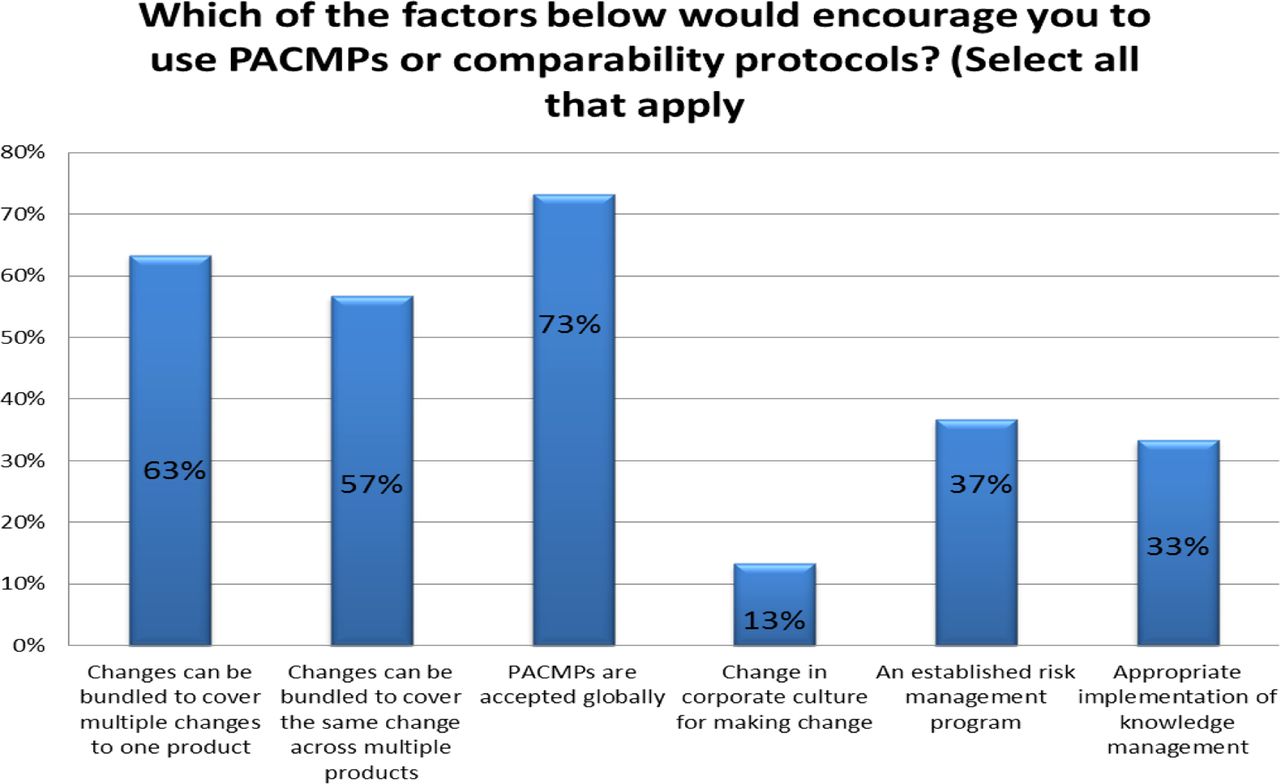
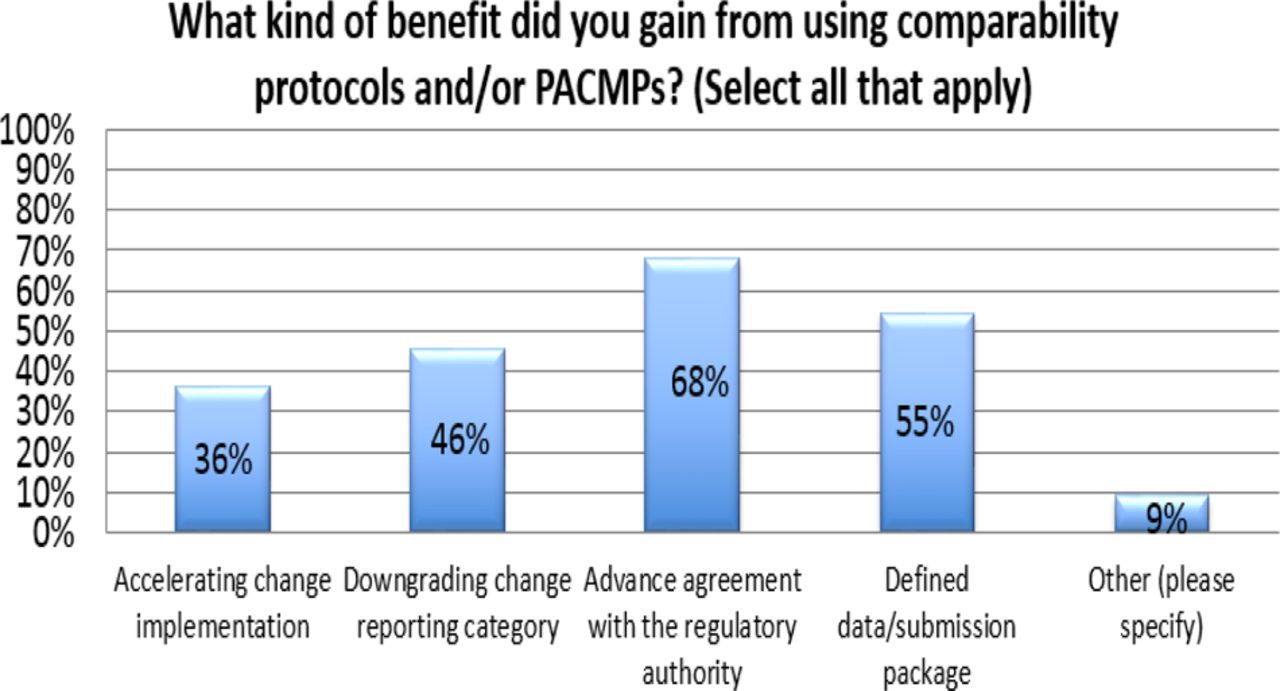
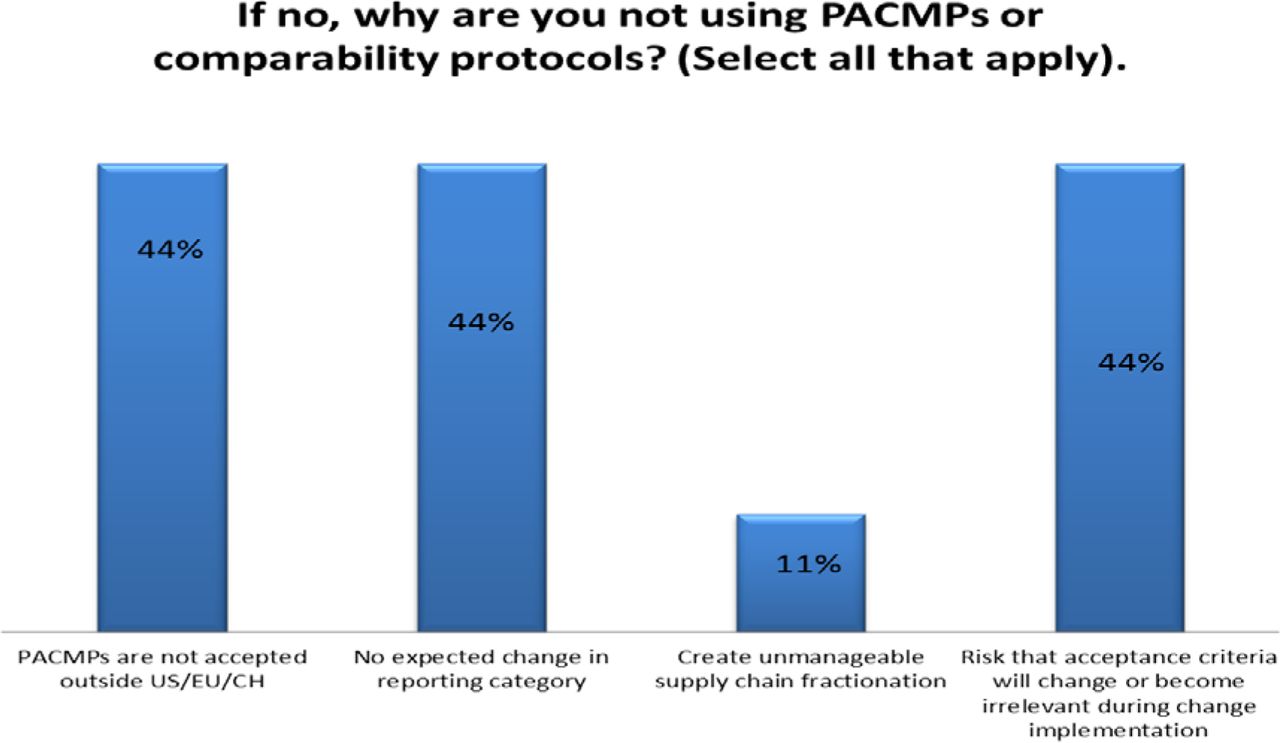
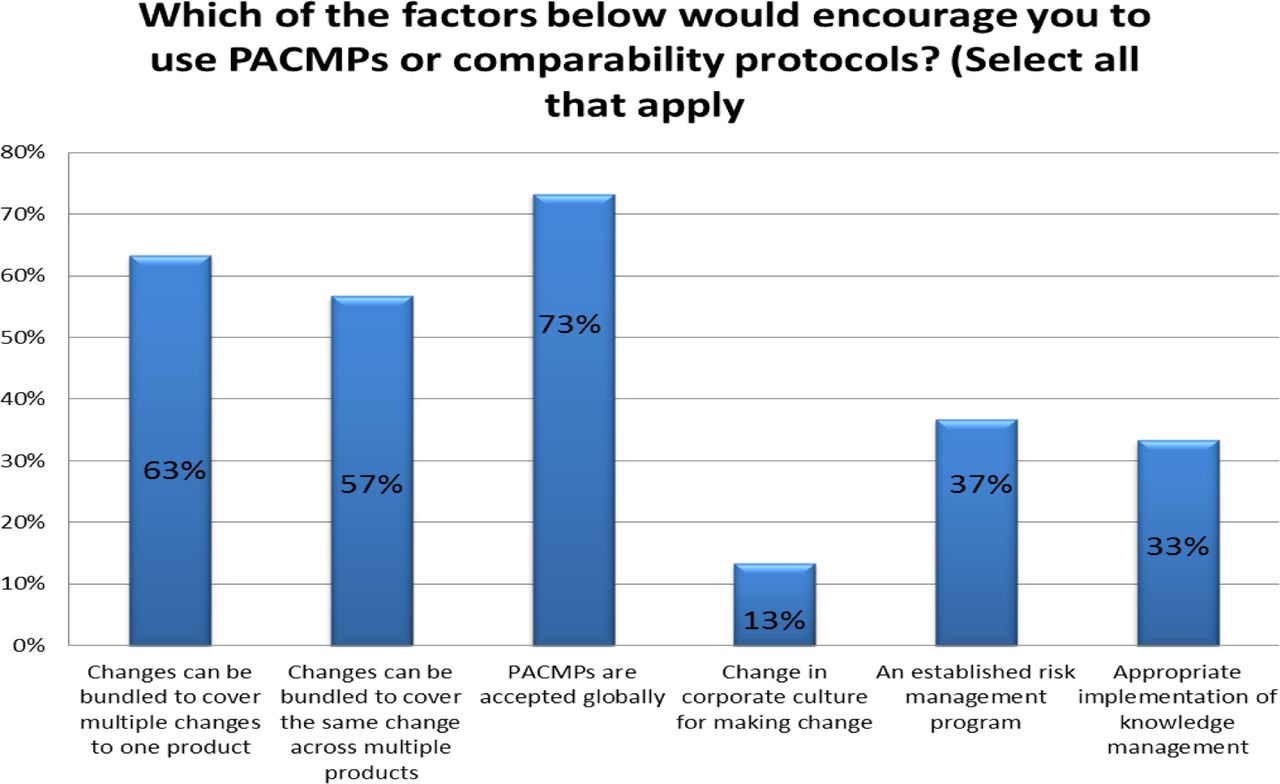
Response from industry indicates that many organizations (73%) are utilizing PACMPs or comparability protocols to lessen the burden of PAC management. This process is allowing companies to accelerate change implementation, advance early communication and agreement with regulators, and in some cases (46%) downgrade the change reporting category (see Figure 10). Unfortunately, some companies (27% of respondents) do not use PACMPs regularly due to lack of acceptance outside US/EU/ICH and a perception that reporting category would not change significantly (see Figure 11). Leveraging tools such as PACMP or comparability protocols globally appears to be a step in the right direction—improvements including bundling of changes global acceptance of PACMPs, and structured approaches for risk management and knowledge management, could drive more uptake and realization of benefits of this tool (see Figure 12).

Impact on Drug Shortage, Compliance, Innovation
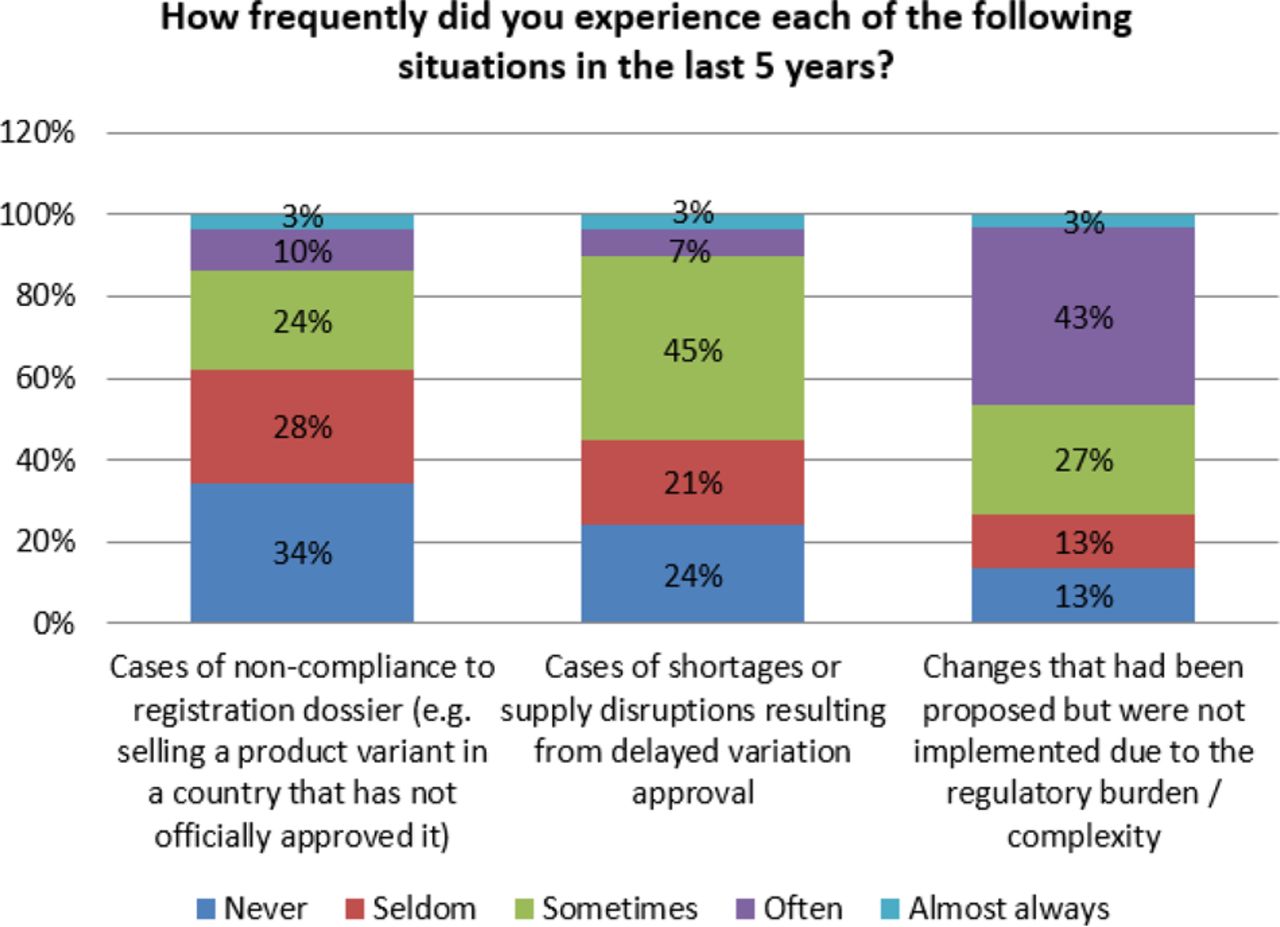
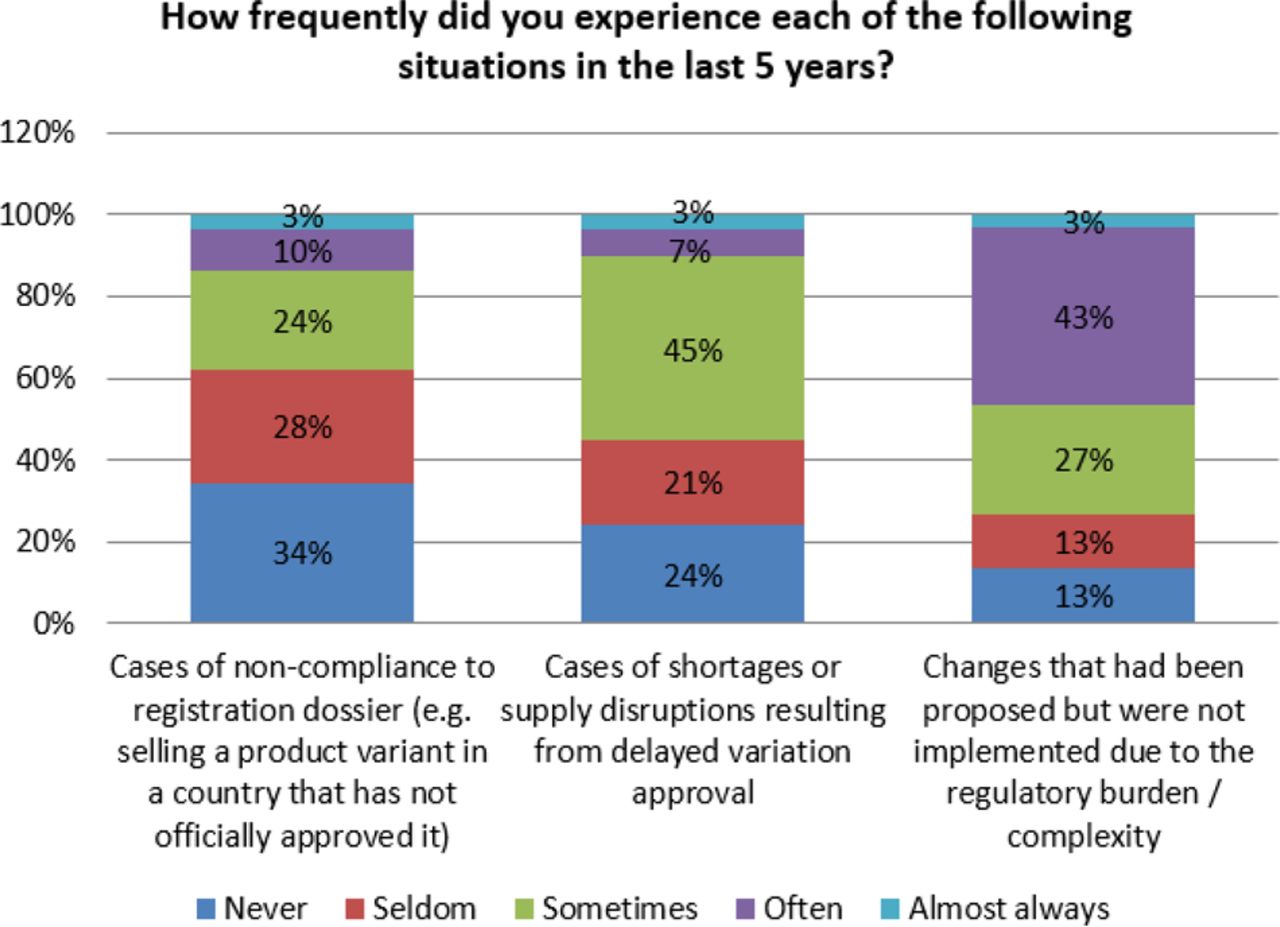
As noted earlier, response from the participating organizations indicates that organizations experience non-compliance and drug shortages due to the complexity of PAC implementation. Seventy six percent (76%) of respondents indicated that they have experienced shortages and supply disruptions due to delayed approvals. Additionally, 65% of organizations reported that they have had cases of non-compliance to their registration due to similar issues. Even more concerning is that 87% of the participants in the survey indicated that changes were not implemented due to the complexity and regulatory burden (see Figure 13). This reluctance to initiate changes based on regulatory burden may influence the industry with respect to innovating in meaningful ways.
The full survey results are available at https://www.pda.org/conference/pac-iam/home.
Conclusions
The 2017 PDA PAC iAM Survey documented the response of industry with respect to the complexity of PAC management. Change approval times can be excessive and many organizations deal with multiple product variants and numerous regulatory submissions, resulting in overly complex supply chains and potential drug shortages and interruptions in supply. The survey also confirms that the impact of the overall complexity of PAC is significant, and not only leads to drug shortages and supply issues but also increases the likelihood of non-compliance to regulatory submissions. While these aspects demonstrate the burden, even more concerning is the indication that many organizations simply forego innovation and process improvements due to this complexity. The complexity of PAC ultimately results in hurdles that are stifling our industry and limiting continual improvement and innovation, where this very innovation can have a significant impact on the health and wellness of millions of patients.
The results of this survey strengthen the mission of the PDA's PAC iAM program as it strives to identify, assess, and address current barriers to implementation of PACs. Recent PDA Points to Consider documents (2, 3) begin to point to possible solutions via PACMPs, leveraging lifecycle management strategies, as well as the use of a company's effective PQS to manage change with minimal or no regulatory reporting. While it is noted that PACMPs and comparability protocols have provided some relief, these solutions are not global and therefore do not address the overall burden leading to a lag in innovation and continual improvement. It is critical for industry and regulators to work together to drive global convergence or regulatory requirements, leverage the company's PQS more effectively to reduce reporting burden, and drive innovation and continual improvement that will ultimately benefit the patients we serve.
Conflict of Interest Statement
The authors declare no conflicts of interest.
Footnotes
PDA PAPER DISCLAIMER: The following paper is a special contribution from the Parenteral Drug Association (PDA). This article was internally reviewed by PDA and the task force members and not peerreviewed by the PDA Journal of Pharmaceutical Science and Technology. Note: This PDA Paper is protected by copyright and unauthorized distribution or use is prohibited.
- © PDA, Inc. 2017




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Introduction
- Summary
- Participation
- Most PACs Reflect the Need to Manage Capacity, Innovate
- Complexity—Timelines and Product Variants Contribute to Inventory Segmentation/Drug Shortage
- Time Required for PACs
- Post-Approval Change Management Protocols (PACMPs) May Help Lessen the Burden
- Impact on Drug Shortage, Compliance, Innovation
- Conclusions
- Conflict of Interest Statement
- Footnotes
- Reference
- Figures & Data
- References
- Info & Metrics