
Abstract
Ophthalmic solutions and suspensions have long been classified into a high-risk category with respect to concerns over extractables and leachables (E&L), though specific guidance on the management of leachables in these products is generally absent from regulatory authorities or the scientific literature. As a result, ophthalmic drug products (ODPs) were originally included in the scope of the Product Quality Research Institute Leachables and Extractables Working Group for Parenteral and Ophthalmic Drug Products (PQRI-PODP). Relative to other high concern dosage forms such as metered-dose inhalers or injectables, ODPs possess unique challenges with respect to the nature of impactful E&L as well as the safety assessment of leachables. For example, extensive use of semipermeable low-density polyethylene primary packaging for ODPs necessitates a strong focus on E&L from secondary packaging sources. For safety assessment, a key challenge is the lack of a sufficient database developed on all relevant ophthalmic toxicity endpoints. As result, the working group is unable to recommend a safety concern threshold (SCT) for ODPs at this time. Nevertheless, the ophthalmic industry has developed a number of time-tested practices to manage E&L for ODPs. This article describes those science-based practices and key considerations in the analysis, management, and safety assessment of E&L in ODPs.
Background
Ophthalmic solutions and suspensions have long been classified into a high-risk category with respect to safety concern over extractables and leachables (E&L) (1, 2). Despite this classification, concrete published guidance on the management of E&L in ophthalmic drug products (ODPs, specifically solutions and suspensions) is absent from the literature. Ophthalmic companies have followed similar practices since the early 2000s, but these practices are also generally absent from the published literature. The goal of this article is to capture E&L challenges specific to ODPs and provide examples of practices successfully implemented by some ODP manufacturers.
In an effort to standardize E&L practices across drug products, ODPs were originally in scope for the Product Quality Research Institute Leachables and Extractables Working Group for Parenteral and Ophthalmic Drug Products (PQRI-PODP). This working group was established to extrapolate the PQRI-Orally Inhaled and Nasal Drug Product (OINDP) E&L threshold concepts and best practices (3) to PODP based on a three-point hypothesis (4, 5):
Threshold concepts that have been developed for safety qualification of leachables in OINDP and the existing U.S. Food and Drug Administration (FDA)/European Medicines Agency (EMA) guidance documents can be extrapolated to the evaluation and safety qualification of packaging systems (such as container closure systems) for PODP.
The good science practices that were developed for the OINDP pharmaceutical development process can be extrapolated to packaging systems for PODP.
Threshold and best practices concepts can be integrated into a comprehensive process for characterizing packaging systems with respect to leachable substances and their associated impact on PODP safety.
Given the hypothesis-driven nature of the PQRI process, the thinking of the PODP Working Group evolved significantly over time. The original OINDP Working Group recommended a safety concern threshold (SCT) of 0.15 µg/day that effectively determined how low the analytical chemist must probe drug product for leachables (3). This conservative value was driven by a number of risk factors exhibited by metered-dose inhalers (MDIs), the most conservative case considered by the OINDP E&L Working Group. These risk factors included the chemical nature of likely E&L from MDI packaging systems, the strong solvents present in MDI formulations that significantly enhance the likelihood of leaching, and the fact that the dose is delivered directly to the diseased organs of a sensitive patient population (3). Because these risk factors are significantly lower for PODP, particularly given a preponderance of aqueous formulations, the team determined that any SCT applied to injectables would be less conservative and ultimately recommended an SCT of 1.5 µg/day (4, 5).
However, the working group realized over time that parenteral and ophthalmic drug products are sufficiently different that they cannot be readily treated in the same manner. For injectable drug products, an SCT could be generated based on similar principles originally used by the OINDP Working Group. Typical ODPs, on the other hand, are dosed topically in small aliquots directly to the eye. Currently there is not a sufficient database developed on all the relevant toxicity endpoints to allow the working group to recommend specific safety thresholds (i.e., sensitization, ocular irritation) for ODPs at this time. Thus, the hypothesis that threshold principles could be extrapolated from OINDP to ophthalmic solutions and suspensions lacked sufficient scientific support to develop a recommendation.
Although specific thresholds cannot be recommended at this time for ODP, this article was developed to capture the challenges specific to ODPs and elements of the second part of the PODP hypothesis, good scientific practices for pharmaceutical development that exist within the ophthalmic industry. For ODPs, low-density polyethylene (LDPE) containers are frequently selected as they provide sterile protection while also possessing mechanical properties that allow effective administration of eye drops. Although LDPE containers readily meet these product requirements, they are also semipermeable to chemical species thereby introducing the potential for leachables derived from secondary (nonproduct contact) packaging. Therefore, it is important to recognize that when semipermeable primary packaging is used, the potential for leachables from secondary packaging needs to be carefully considered. Many good science practices exist to manage the issues arising from the prevalence of semipermeable LDPE primary packaging and the resulting susceptibility of ODPs to leachables from secondary packaging. It is the objective of this article to capture these practices along with strategies for safety assessment.
Importance of Assessing Leachables in ODPs
A large percentage of ODP dosage forms are solutions and suspensions (including emulsions). Although other dosage forms exist in ophthalmology (e.g., ointments, injectables, implants), the scope of the following discussion is limited to topical solutions and suspensions. Because ODPs are liquid-based, they are more likely than solid dosage forms to interact with and possibly extract leachables from the primary packaging system (e.g., LDPE multidose bottles or unit dose vials). In addition, compared with some other organs (e.g., the gastrointestinal [GI] tract), the eye and surrounding area are generally more sensitive to compounds such as sensitizers or irritants if they are present as leachables. For these reasons, ophthalmic solutions and suspensions are classified to pose a “high” degree of concern in the FDA Guidance for Industry: Container Closure Systems for Packaging Human Drugs and Biologics (May 1999) and USP <1664> (1, 2). Therefore, it is important to conduct extractable and leachable studies to understand the leachable profiles in these products, assess the risks associated with potential and observed leachables, and control the leachable profile as necessary.
It should be noted that in the FDA Guidance (1), although ophthalmic solutions and suspensions are classified in the “high” degree of concern category, they are different from injections (e.g., small- or large-volume parenteral [SVP, LVP] products) that are in the “highest” degree of concern category. Although the risk for leachables in topical ODPs is generally lower than that for injectables, Section III.E. of the document notes that they are often grouped together because they “share the common attributes that they are generally solutions, emulsions, or suspensions, and are all required to be sterile” (1). However, injections and ODPs are different in route of administration (i.e., systemic vs. topical, respectively); thus, the toxicological implications of leachables in these products are also different (e.g., systemic exposure vs. local irritation). In addition, the dosing volumes for injections, especially LVPs versus ODPs, are very different. Therefore, ODPs should be in a different category from injections when performing toxicological evaluation of leachables, thus warranting different considerations (e.g., potential for systemic exposure, toxicity endpoints, concentration- vs. total daily intake [TDI]-based reporting).
Current Regulatory Approaches
Currently there is no official regulatory document providing specific guidance on ODPs from the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) or any of the Health Authorities from the major geographies. The ICH guideline Q3B, Section 1.3 states “Impurities arising from excipients present in the new drug product or extracted or leached from the container closure system are not covered by this guideline” (6). Although the FDA 1999 packaging guidance discusses ODPs, its recommendations regarding them are obsolete versus current expectations. For example, for Case 2s products such as ophthalmic solutions and suspensions (as well as injectables), “typically provided are USP Biological Reactivity Test data and possibly extraction/toxicological evaluation.” Clearly, this guidance understates the current FDA expectations around understanding extractables and leachables in ODPs. The EMA published a Guideline on Plastic Immediate Packaging Materials in 2005 (7). The guideline established an expectation that extraction studies are necessary for plastic materials used for container systems of nonsolid active substances and nonsolid dosage forms. The guideline specifically identifies ODPs as in scope but does not provide guidance on reporting, identification, and safety qualification thresholds for leachables. Although packaging is not in scope, the ICH M7 guidance on Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk (2018) does state that “Application of this guideline to leachables associated with drug product packaging is not intended, but the safety risk assessment principles outlined in this guideline for limiting potential carcinogenic risk can be used if warranted”. The M7 document does provide guidance on risk assessment for carcinogenic endpoints but does not provide guidance on assessment of other safety endpoints (8).
In February 2011, at the PQRI workshop on Thresholds and Best Practices for Parenteral and Ophthalmic Drug Products, a representative from the FDA’s Office of New Drug Quality Assessment (within the Center for Drug Evaluation and Research [CDER]) made a presentation that covered leachables for ophthalmic solutions, suspensions, and emulsions in LDPE container closure systems (9). The presentation discussed principles of applying a science-based approach to study design, format of reporting leachable results, and thresholds for reporting, identification, and qualification of leachables. The presentation recommended reporting leachables results in an absolute concentration format such as parts per million (i.e., microgram per milliliter or microgram per gram) rather than as a weight percentage of the active ingredient. The presentation also recommended that the thresholds of reporting, identification, and qualification be 1, 10, and 20 ppm, respectively. In practice, the reporting of leachables exceeding 1 ppm is commonly done by ophthalmic companies; however, most Sponsors identify below 10 ppm and provide available safety data for each identified leachable such that the 10 and 20 ppm values are less relevant. The extent of additional data that is required is considered on a case-by-case basis. Rationales for basing thresholds on concentration rather than on daily exposure, and choosing the numerical threshold values were not provided. Note that, although the content of this presentation has been in the public domain, it has not been captured in any official FDA guidance. The aforementioned thresholds on reporting, identification, and qualification are not official FDA guidelines. They are described here as a reference to provide insight into a possible approach or starting point, but not as a practice that must be followed.
In the following section, we present recommendations, with experiential justification, on concentration-based leachable reporting for ODPs. It is important to note that some of these recommendations differ from those given for parenteral drug products, specifically in that no safety-based threshold is recommended for ODPs at the current time. The toxicological considerations and justifications are discussed along with particular analytical considerations for ODPs.
Specific Considerations for Managing Leachables in ODPs
For topical ODPs, typically low drug amounts (<10 mg) in small volumes (<100 µL, often 30–50 µL) are administered to the eye of the patient. As compared with LVPs, the low volume of solution contained in each bottle could result in a relatively high concentration of leachable in the final product. Unlike process and degradation impurities, which are reported as percent of the drug substance, leachables are not related to the drug and therefore should be reported as an absolute concentration (weight of leachable per unit volume or mass of drug product) and not as a weight percentage of the active pharmaceutical ingredient (API). Additionally, as ODPs are topical solutions/suspensions, it is recommended to report levels of leachables in terms of concentration (parts per million) and not TDI, as this measurement is more relevant to the toxicological evaluation of local effects on the eye, which are of primary concern for this product type. However, when considering other endpoints such as genotoxicity (which is primarily a systemic toxicity concern) as part of toxicological qualification for ODPs, it can be useful to also calculate the TDI or the maximum daily exposure to include as another means of evaluation in the safety assessment (i.e., comparison to the threshold of toxicological concern [TTC] level for genotoxicity).
Unlike the recommendation for ODPs, reporting and assessment of leachables for other product types such as parenterals or OINDP are based on thresholds established based on TDI. For comparative purposes across product types, Table I illustrates for leachables the relationship between TDI (comparison at parenteral product SCT levels) and concentration on eye (parts per million) as a function of dosing frequency and number of eyes treated. Note that at a constant TDI, the concentration of a leachable on the eye varies and is inversely proportional to the number of daily doses. It is recognized that for a once-daily-dosing ophthalmic product, with such a small volume of administration, the calculated leachable concentration could be quite high at a TDI considered fairly low (15 ppm for a TDI of 1.5 µg for a twice daily product), supporting the recommendation that a concentration-based approach is more appropriate for this product type for which local effects on the eye are of primary consideration.
Comparison of Total Daily Intake to Concentration Values: Relationship to Dosing Frequency
Ocular drugs are designed to achieve a high local exposure and corresponding low systemic exposure. As lower amounts of drug are needed to achieve a local effective concentration and only typically 50%–80% of the topically administered drug is absorbed systemically, systemic exposure (peak concentration) is often in the low nanogram per milliliter range (10).
Therefore, for ODPs, local topical (ocular) effects are considered a primary area of concern for toxicity potential. Systemic effects including general and developmental and reproductive toxicity are less relevant to ODPs relative to other product types. For ODPs, safety thresholds and exposure for local effects (i.e., sensitization and irritation/toxicity) will be a primary consideration when qualifying leachables and extractables. However, consistent with qualification of other impurities and starting materials, and with the knowledge that ophthalmic drugs may be used for both acute and chronic duration indications, genotoxicity potential is also considered a relevant endpoint for this product type.
Therefore, the primary toxicological endpoints of consideration for qualifying leachables for topical ophthalmic products include ocular irritation and toxicity, sensitization, and genotoxicity.
Potential Sources and Common Classes of Leachables for ODPs
With respect to extractables and leachables (E&L), each therapeutic area and/or dosage form is characterized by specific challenges. A key challenge for ODPs arises from the prevalent use of semipermeable LDPE multiuse bottles or unit dose vials as primary packaging. The primary packaging is an obvious source of potential leachables because of its direct contact with the dosage form. In addition, because LDPE containers are semipermeable, compounds from components in direct contact with the outside of the bottle (such as label adhesive and ink printed on the bottle) can migrate through the bottle into the dosage form. Although the USP differentiates leachables as those compounds that migrate from direct contact primary packaging from migrants that must cross a physical barrier to enter the drug product (2), both classes of compounds will be described as leachables for the purpose of this discussion.
Furthermore, experience within the ophthalmic industry demonstrates semivolatile or volatile compounds from other secondary packaging systems (e.g., unit carton, foil pouch, labels, and product information inserts) can migrate through the semipermeable primary packaging system into the dosage form. Such volatile compounds include components of carton paperboard, foil pouches, adhesives, and inks printed on the label or product information insert. When the primary container is packaged in sealed, impermeable secondary packaging (e.g., a foil laminate pouch used to protect the primary container from losing water), migration of leachables from secondary packaging components within the pouch becomes especially significant. In this case, because volatile leachables from the inside of the secondary packaging system cannot escape to the outside environment, their equilibrium concentrations in drug product may be elevated. Therefore, consideration of leachables from sources outside of the semipermeable primary packaging system is critical during leachable studies.
Common classes of leachables in ODPs include residual monomers or low-molecular-weight oligomers and additives from the primary packaging material. Additives may include antioxidants, light stabilizers, plasticizers, colorants, antistatic agents, and lubricants or mold release/slip agents. Historically, ODP manufacturers have been challenged to manage leachables from secondary component sources. General examples include phenolic unit carton paperboard preservatives, photoinitiators, and other compounds from inks and coatings on labels and unit cartons (11), surfactants from label adhesives, low-molecular-weight plasticizers from unit carton components, and many others.
Leachables from secondary packaging are not entirely unique to ODPs because other therapeutic areas also make use of semipermeable primary containers, but they are particularly prominent for ODPs. Because these secondary packaging components do not make direct contact with the drug product and, in some cases, only make incidental contact with the primary container itself (e.g., unit cartons), many of the observed leachables are low-molecular-weight compounds with sufficient vapor pressure that they can be transported through the air (12). As with all generalizations, there are exceptions, however. For example, polymeric surfactant species (molecular weight 400–700) have been observed to migrate into drug products from label adhesives in direct contact with the primary container. Not surprisingly, the rates of migration for such compounds are significantly less than those for low-molecular-weight leachables. Thus, researchers studying ODPs should not restrict their attention exclusively to small, volatile or semivolatile molecules. Furthermore, the example of the polymeric surfactant highlights the idea that different compounds migrate at different rates.
False Negatives Observed during Extractable Testing of Secondary Packaging Components and Leachable Testing of Drug Products for Which Secondary Packaging Components Are Critical
Because many leachables originating from secondary packaging are volatile or semivolatile compounds, the analytical chemist who seeks to characterize them is faced with additional challenges. Care must always be taken in sampling secondary packaging components to ensure that the integrity of the sample is preserved. For example, consider a case study of a phenolic preservative from unit carton paperboard that migrated into a drug product. During extractable testing, an individual sheet of the paperboard mailed in a paper envelope to an analytical lab on the other side of the country is likely to arrive for analysis significantly depleted in volatiles or semivolatiles, such as the preservative of interest. Extraction testing of such a material may generate false-negative results for likely leachables, which would undermine the objective of extractable testing. Therefore, sampling and shipping of secondary components for any extractable testing must always be done in a manner that minimizes loss of key extractables. Although this is important for any analysis, it is critical for secondary packaging because the compounds that are most likely to be lost in transit are also those that are likely to migrate into a drug product.
In addition to extraction studies, false-negative results have been observed to occur as a function of storage temperature in long-term leachable studies. Drug product leachables are often analyzed in parallel with registration stability. These long-term studies, typically run in real time for the duration of the intended shelf life of the product, carry an intrinsic risk that a problematic leachable will be discovered late in the study. As a result, there is often a desire to accelerate the leachable study to assess risk earlier in the drug product development program. However, experience with secondary packaging leachables indicates that accelerated studies are rarely predictive of long-term, real-time leachable levels. Empirically, accelerated stability studies run on products in semipermeable containers frequently underreport key secondary packaging leachables versus real-time data. Figure 1 provides an example comparison of a unit carton leachable tracked in an ODP filled into LDPE at real-time and accelerated conditions. Note that the accelerated condition underestimates the real-time end of shelf life leachable concentration by a factor of 2. This phenomenon has been observed by multiple ODP manufacturers. Though it is directionally consistent, the magnitude may vary. In some more extreme instances, a drug product stored at a higher (accelerated) temperature will appear to be free of detectable leachables, whereas reportable levels of leachables are observed after storage at the lower temperature real-time condition. However, ODPs encapsulated in impermeable secondary packaging (e.g., foil laminate pouches) are not susceptible to this effect because any volatile or semivolatile extractables evolved from components within the pouch are trapped in close proximity to the dosage form and are unlikely to be lost to the wider environment.
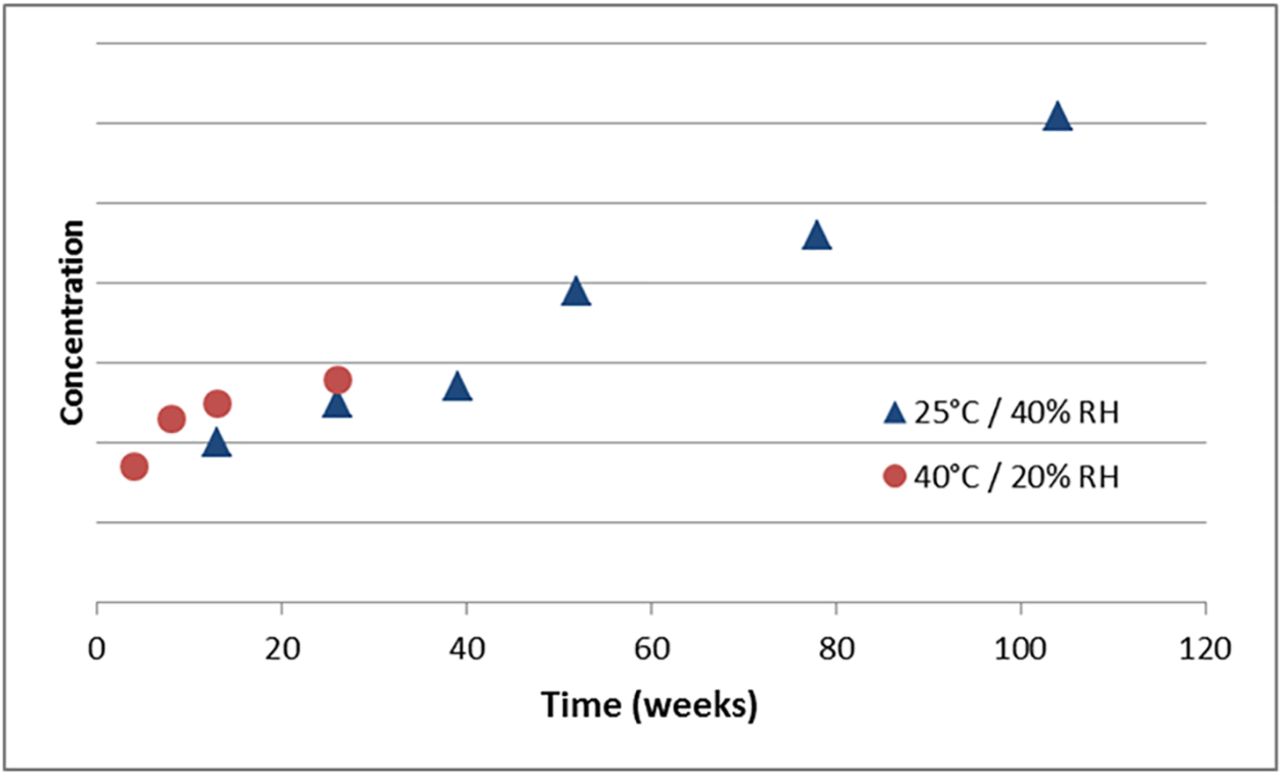
Impact of storage temperature on concentration profiles of secondary packaging leachables. Example comparison of real-time and accelerated data for a unit carton leachable observed in the same lot of ODP with an LDPE primary container. In this case, the product is not encapsulated in a foil pouch. LDPE, low-density polyethylene; ODP, ophthalmic drug product; RH, relative humidity.
The concept of accelerating chemical degradation is well established through the use of an Arrhenius relationship that either assumes or empirically derives an activation energy appropriate to the chemical reaction under study. However, it is important to remember that leaching is not a chemical process like oxidation or hydrolysis. Rather, it is a partitioning phenomenon with a rate that will be impacted by many variables such as: the source of the leachable (product contact or nonproduct contact components), leachable mobility (as governed by its individual physicochemical properties), properties of the other packaging components in the system (such as the barrier properties of the primary container), and the local environment of the storage chamber (which can influence the equilibrium levels and ultimate sink for volatile or semivolatile leachables). Thus, although an Arrhenius model may be used to derive an apparent activation energy for the leaching of a specific compound from a secondary packaging component, the assumption that all leachables will obey the same concentration profile over time or possess the same apparent activation energy is demonstrably false. Experience within the industry explicitly demonstrates that leaching rates can vary significantly between specific leachables. Therefore, these compounds cannot be expected to obey a common model. As a result, acceleration of leachable studies in which secondary packaging is critical must be treated cautiously with the awareness that relating accelerated data to real-time behavior may be challenging. Strategies for managing the longer-term risk of a reportable leachable being discovered late in the development process as well as avoiding false negatives will be discussed in a later section.
In conclusion, the wide use of semipermeable primary packaging systems for aqueous ODPs is generally the root cause for most challenges affecting these products, requiring that significant focus be placed on secondary packaging components. Fortunately, these challenges can be mitigated by proper experimental design.
Experimental Design Considerations: Extraction/Simulation Studies for ODPs
Analytical laboratory work often begins with a controlled extraction study. As recommended by the PQRI-PODP Working Group (3), “controlled extraction studies should use a combination of multiple relevant extraction solvents of varying chemical nature and suitable extraction techniques as appropriate for, and consistent with, the intent and purpose of the controlled extraction study.”
It is important to realize that extraction studies can be performed to satisfy a variety of purposes. As with any scientific endeavor, the experimental design may vary as a function of the question that the study is intended to address. For example, extraction studies may be performed for the purposes of material characterization (what compounds can be withdrawn from a particular material or component?), simulation (which compounds are probable leachables for a given dosage form and packaging system combination?), quality control (are incoming batches of components or materials consistent with historical batches?), or change control (what is the impact of changes in process or material to a component in the packaging system?). Experimental design and, particularly, solvent choice must be aligned with the purpose of the study.
Extraction studies conducted for purposes of material characterization generally require the most aggressive extraction conditions and/or solvents. These studies are intended to impart comprehensive understanding of the compounds that can be withdrawn from individual, specific packaging components. Given that most ODPs are highly aqueous, water must be a prominent solvent in these extraction studies, and the effect of pH is of particular interest. As described previously, pH may be a relevant variable based on the nature of the product(s) intended for the packaging system (13). Many ODP formulations contain polarity modifiers (e.g., demulcents, surfactants, preservatives such as benzalkonium chloride, and so forth,) such that they are better sinks for leachables than straight water or simple buffered aqueous formulations. With extractable testing, the researcher has significant latitude in solvent choice based on the system under study. Given that the objective of material characterization is to obtain greater knowledge of extractable compounds from individual components, stronger solvents such as 2-propanol (or mixed 2-propanol/water solvents) are recommended for inclusion in the study (13). 2-Propanol is useful for extraction of ODP packaging components; it is more aggressive than most aqueous ODP formulations, but less aggressive than methylene chloride and hexane. Furthermore, it is a particularly good solvent for extracting the phenolic antioxidants commonly found in polyolefins (13). Given the preponderance of polyolefinic materials used for ODP primary packaging components, 2-propanol extraction data can be quite useful at the onset of an E&L study. A sound understanding of the additives from primary packaging components can also impart understanding about their degradation or transformation products, many of which are well-known in the literature (14, 15). Because these transformation products tend to possess lower molecular weight (e.g., the hydrolysis products of Irganox 1010) and/or greater polarity (e.g., the oxidized phosphate form of the phosphite antioxidant Irgafos 168) than the precursor additives, they are more likely to appear as leachables in aqueous drug products.
In many ways, application of extractable testing for purposes of material characterization of ODP primary packaging components is well aligned with the general design and execution of extraction studies for other drug product types (3, 5). However, the challenges intrinsic to secondary packaging when combined with semipermeable container closure systems warrant a refined approach. In this arena, simulation studies possess significant value.
Though still a variation on extraction, simulation studies are focused on answering a different question than material characterization. Whereas material characterization strives for comprehensive understanding of extractables from individual packaging components, simulation studies are intended to forecast the most probable leachables for a specific combination of packaging system and drug product.
Simulation study designs are quite varied but generally possess a few common characteristics. First, simulation studies tend to use product-like solvents (including placebos, where practicable). By approximating the propensity of leachables to move into the drug product of interest, simulation studies are more likely to return quantitatively relevant data that help researchers focus on the most important extractables. Furthermore, use of a simple solvent system for simulation in lieu of drug product holds the potential to facilitate the detection and identification of probable leachables in a less complex matrix.
Another characteristic of simulation studies is that they can monitor packaging systems, or relevant portions of packaging systems, under realistic contact conditions rather than by direct solvent extraction of individual nonproduct contact components. By designing simulation studies using realistic packaging component contact conditions, the relevance of observed extractables is further increased. In fact, simulation studies bring focus to probable leachables in ways that traditional extraction studies focusing on individual components (e.g., material characterization) do not. This capability is particularly useful for ODPs and other drug products for which leachables from secondary packaging components are a concern.
For example, consider a label placed on the exterior of an LDPE bottle containing an aqueous ODP. A direct solvent extraction of the label, particularly a strong solvent irrelevant to the drug product such as hexane, will generate a significant number of extractables (potential leachables). Practical experience demonstrates that not all of these targets will appear in the drug product as actual leachables. Furthermore, although direct solvent extraction is meaningful and relevant for primary packaging components that make direct contact with theliquid formulation, it is not realistic for secondary components such as a label. Direct solvent extraction of a label provides a vector for substance migration that simply does not exist in the drug product.
The unrealistic nature of direct solvent extraction for secondary packaging components is further exaggerated by the fact that the hypothetical label in this example represents a variety of different components, each characterized by their own sets of unique extractables. For example, a label may possess a polyolefin substrate, meaning that it is likely to contain the same sort of antioxidants and other additives (along with their associated transformation products) commonly observed from polyolefin primary packaging. Contributing further to label extractables are the adhesive, the ink, and the varnish; all of which may be characterized as complex formulations that may possess unique chemistries from each other. Thus, the diversity of extractables removed from a label can be quite significant. An unrealistic contact condition (direct solvent extraction) combined with a wide variety of potential extractables can lead to generation of complex extraction profiles. If the goal of the extraction study is to choose targets for a follow-up leachable study, the researcher is instead left with a large number of mostly irrelevant targets. Ideally, an extraction study for a label would generate data that focus on the most relevant extractables (i.e., the most probable leachables). This is something that simulation studies accomplish very well.
One technique that is often of value for examining probable volatile leachables from secondary packaging is headspace gas chromatography. The process of measuring analytes in the headspace mimics the mechanism of migration from the secondary component to the drug product (e.g., transport through the air). In this regard, headspace makes an interesting screening tool. However, a more refined simulation study design is discussed following.
In order for a compound from a secondary packaging component to become a leachable, it must satisfy three conditions. First, it must be able to migrate from the original component. Consider a unit carton extractable possessing no significant vapor pressure. Given that unit carton extractables must pass through the air to reach the primary container, an extractable with insufficient vapor pressure to transfer in this fashion will be unlikely to manifest as a leachable. Second, the compound must be able to permeate the primary container. Despite its well-known semipermeable properties, LDPE does not pass all compounds. Rather, the primary container serves as a selectivity element within the packaging system. Thus, compounds that cannot permeate the bottle are not probable leachables. Finally, the potential leachable must dissolve in the formulation. Given that leachables are often probed at low part-per-million or high part-per-billion levels, this latter point is not a significant condition to be met because high solubility is not required. Interestingly, this last point is the only one of the three conditions that is specific to the drug product formulation.
Probable leachables can be determined by probing the properties of the packaging system and its associated extractables in a simulation study using a realistic solvent and realistic contact conditions. Thus, instead of subjecting a label for an aqueous ODP to an unrealistic reflux in a strong solvent, a suitable simulating solvent could be filled into the primary container with a label applied to the exterior as it would be on a finished drug product. Thus, the physicochemical properties of individual extractables as well as the selectivity imparted by the packaging system can be leveraged to identify those compounds most capable of migration into the drug product.
As noted earlier, long-term leachable studies carry the inherent risk that a problematic leachable will be discovered too late in development for facile management. Accelerated leachable studies are often proposed to mitigate this risk and provide data to development teams in a timelier manner. However, accelerated leachable studies on packaging systems with critical secondary components can be poorly predictive and may lead to false-negative results if the study is not properly designed. Such outcomes do little to manage risk from leachables observed late in the leachable study. However, an appropriately designed simulation study may serve to reduce this risk.
Aside from the physical construction of the packaging system, key parameters of most simulation studies include solvent, storage temperature, and study duration. A suitable simulating solvent should be product-like, though skewing toward a more aggressive solvent system will reduce the risk of false negatives (i.e., failure to detect a relevant extractable). The challenge in designing simulation studies is that they should skew sufficiently worst case to credibly avoid false negatives without being so aggressive that they generate too many false positives. This requires individual researchers to design experiments that balance the risks appropriately for the system under study; there is no one-size-fits-all approach to be recommended here.
As described earlier, higher (accelerated) storage temperatures can result in false negatives with respect to leachables from secondary packaging components. The final equilibrium concentration of a volatile or semivolatile extractable/leachable in a solvent/drug product can be influenced by the immediate environment of the packaging system. If the system (e.g., filled product bottle, label, and unit carton) is contained within an impermeable vessel of limited volume (e.g., a well-sealed glass jar or foil laminate pouch), the resulting equilibrium will favor the solvent/drug product because any volatile or semivolatile extractables will be prevented from dissipating into the broader environment. This is directly analogous to the effect discussed earlier in which the concentrations of leachables from secondary packaging contained within a foil laminate pouch are enhanced. In this configuration, storage temperature can be increased to expedite migration without leading to false-negative results. If anything, such a configuration provides a relatively worst-case measure of those compounds capable of migrating from their source component, permeating the bottle, and dissolving in the simulation solvent. If simulation (or leachable) study test articles are enclosed in a sufficiently impermeable container, ICH accelerated storage temperatures or higher (e.g., 60°C) may be used without fear of false negatives. It must be emphasized that no model exists to correlate the levels observed in such a study with real-time data. However, such studies can often provide worst-case equilibrium concentrations of probable leachables in a reduced timeframe to aid in managing risk for a long-term leachable study. As a final note on elevated temperature, it is important to choose a temperature that does not cause significant physical changes to the primary container closure system. For example, storage temperatures higher than 60°C are likely to cause physical deformation of a typical ODP container molded from LDPE.
The final key variable in this sort of simulation study is duration. Running such a study until the system reaches equilibrium will provide a limiting value for the concentration of each potential leachable. In our experience, equilibrium levels of some volatile or semivolatile extractables in elevated-temperature simulation studies may be reached within 1 to 2 weeks. The appropriate study duration for the target compounds can be determined by testing at multiple time intervals and noting when the target compounds reach asymptotically limiting concentrations.
It is important to recall that a simulation study is just that—a simulation. A long-term leachable study in the ODP of interest may still be required as confirmation. It is also important that simulation studies skew slightly worst case with the goal of avoiding false negatives. This comes at the risk of generating false positives and it is up to individual scientists to determine how to best balance those risks. By now, it should be clear that the parameters of a simulation study can be flexible as required to answer specific scientific questions. Careful thought and good science must be applied to the design of simulation studies to produce an experiment that provides scientifically meaningful data and is credible to regulators. Although simulation studies are not necessarily simple, one-size-fits-all experiments, their value is derived in part from their flexible nature.
Thus, simulation studies possess significant value for ODPs or any dosage form for which understanding the impact of secondary packaging components on the drug product leachable profiles is critical. In many cases, simulation studies can serve to identify the most relevant extractables (e.g., the most probable leachables) as targets for long-term leachable studies more capably than a direct solvent extraction. By designing a study with realistic packaging component contact conditions, the experiment provides selectivity for the most probable leachables. Although simulation studies usually run for a longer duration than direct solvent extractions, they are substantially shorter in duration than a real-time leachable study (weeks versus months). Thus, simulation studies can provide earlier understanding of key probable leachables. By providing this information in a timely manner, simulation studies reduce risk during product development such that long-term leachable studies become confirmatory rather than exploratory.
Experimental Design Considerations: Leachable Studies for ODPs
In their execution, leachable studies on ODPs do not vary significantly from those described for other therapeutic areas and/or dosage forms. Fundamentally, well-designed leachable studies are the most conclusive demonstration of packing system suitability for a given ODP but also require the greatest time investment.
It is expected that leachable studies will run in real time through the end of shelf life. This is typically done in parallel with registration stability. Inclusion of key secondary packaging components (e.g., label, product information insert, unit carton, and so forth) in the study is critical for ODPs in semipermeable primary packaging containers (e.g., LDPE). Health authorities are quite aware of the challenges posed by semipermeable packaging systems (1) and will expect sponsors to develop understanding of the impact that secondary packaging components have on the drug product. As described previously, accelerated stability conditions tend to generate nonpredictive or false-negative data for secondary packaging leachables unless measures are taken to manage the evaporative loss of these substances from the local environment. For products to be offered in multiple fill volumes, those combinations presenting the highest ratio of secondary component mass (which generally scales with surface area) to product fill volume tend to generate worst-case leachable levels over time (barring extenuating variables like significant changes to bottle material type or bottle wall thickness).
Because of the large variety of potential secondary packaging leachables, it is often useful to run suitable negative controls in parallel with the leachable study. Typically, such controls are implemented by filling bulk drug product into clean, impermeable containers. Glass vials with Teflon-faced screw cap closures are often useful for this purpose. If the bulk drug product has not been previously exposed to the packaging system (primary or secondary), comparative analysis between the negative controls and packaged drug product readily serves to differentiate leachables from other analytical responses associated with the formulation or its degradation over time. This approach has been a standard practice within the ophthalmic industry for many years.
Analytical Evaluation Threshold
Key concepts introduced by PQRI in 2006 (3) and expanded upon by the PODP Working Group (5) include the SCT (0.15 µg/day for OINDP, 1.5 µg/day for injectables) and the analytical evaluation threshold (AET). The AET is a reporting and identification threshold that represents the SCT corrected for dosing of the specific product. Together, the SCT/AET concepts provide the analytical chemist a target reporting threshold for unknown leachables. This threshold is derived from a safety model and based on total daily exposure for products dosed in a manner that facilitates systemic exposure.
Currently there is not a sufficient database developed on all the relevant toxicity endpoints to allow the working group to recommend specific safety thresholds for ODPs. Thus, the parenteral SCT of 1.5 µg/day introduced for parenteral products by the PQRI-PODP Working Group cannot be applied directly to ODPs (4, 5). Ophthalmic manufacturers have successfully used concentration-based, as opposed to exposure-derived, reporting thresholds for many years (9), although no official guidance is available.
Summary of Analytical Challenges for Extractables and Leachables in ODPs
In summary, many of the analytical challenges for ODPs result from the common use of semipermeable primary container closure systems. In these systems, appropriate use of well-designed simulation studies can be leveraged to focus on the most important extractables from critical packaging components and allow for relatively rapid long-term risk assessment. Overall, the above measures are part of a holistic approach to minimize the presence of leachables in ODPs as described below:
Understand the compositions of the packaging materials (including both primary and secondary packaging) by obtaining information from the suppliers of the materials. Based on supplier information, a list of potential extractables/leachables may be compiled.
Conduct extractable/leachable studies on both primary and secondary packaging to evaluate the levels of the leachables. As described previously, simulation studies often work well in lieu of direct solvent extraction for profiling secondary packaging components.
If certain leachables are determined to be present in the dosage form at considerable levels, the toxicological effects of these leachables should be evaluated.
Based on the preceding evaluation, select the proper packaging materials from the appropriate suppliers to limit and control the leachables at acceptable levels.
Toxicological Considerations for Leachables in ODPs
The primary toxicological endpoints that need to be considered for qualifying leachables for topical ophthalmic products include (i) ocular irritation and toxicity; (ii) sensitization (skin), and (iii) genotoxicity. A rationale and specific considerations for each endpoint when qualifying a leachable are outlined next.
For topical ODP leachables that are not genotoxicants or carcinogens, because of the local concentrated delivery of the drug, ocular irritation/toxicity is a key toxicity endpoint. The establishment of a qualification threshold for ocular irritation/toxicity would be useful as it would help overcome the current difficulty in adequately assessing the safety of a leachable via the literature (because of the lack of available relevant ocular data) and decrease the need for unnecessary animal testing. There are currently no official regulatory guidance documents that define a threshold level for ocular irritation/toxicity, and there is not a sufficient database developed on ocular irritation/toxicity for this working group to recommend one at this time.
However, the FDA’s Office of New Drug Quality Assessment previously presented a loosely followed practice for managing the presence of leachables within ODPs, and within this presentation it was suggested that the toxicological qualification level for leachables was 20 ppm (9). Although this is not considered an FDA-endorsed practice, it does provide a starting point when attempting to establish a rationale-based threshold for ocular endpoints through literature and/or experimentally derived data.
Preliminary studies have been conducted to evaluate the potential of establishing a threshold for ocular irritation/toxicity. In these studies, 11 chemicals from 9 different classes (acids, acrylates, acyl halides, alcohols, alkalis, amines, and surfactants) were tested in New Zealand White rabbits for ocular irritancy potential in 3-day multidose studies at concentrations of 20 ppm and 100 ppm (50 μL dose). The five rabbits/compound were dosed four or six times (depending on the vehicle used) each of the 3 days. When possible, the vehicle was phosphate-buffered saline (PBS) and dosing was 6×/day. For chemicals insoluble in PBS, cottonseed oil was used as the vehicle, and dosing was limited to 4×/day to avoid potential vehicle-related effects. The rabbits’ eyes were examined daily macroscopically using modified Draize scoring before the first dose and after the last daily dose. The animals were also examined and scored via the McDonald-Shadduck method using a slit lamp on Day 3 following the final Draize evaluation. For all compounds and at both 20 and 100 ppm concentrations, there was no evidence of eye irritation when macroscopically examined and scored using the modified Draize method or upon slit-lamp evaluation. All animals appeared clinically normal throughout the duration of the study.
The favorable ocular irritation results observed with up to 100 ppm concentrations of the selected severe irritants suggest that it may be possible to establish a threshold for ocular irritation/toxicity. Each chemical under evaluation is a severe ocular irritant with the potential to cause serious damage to the eye at concentrations much higher than expected levels of leachables in ophthalmic formulations. That these chemicals showed no evidence of irritation in the rabbit eye at 100 ppm supports that 20 ppm may be in the appropriate range as a potential threshold for ocular toxicity endpoints. However, further work using a greater number of compounds and evaluating additional toxicity endpoints is needed to verify a specific threshold value.
For considerations related to sensitization, the eye is generally considered immune-privileged, and as such, is largely protected by the blood–retinal and blood–aqueous barriers from systemic induction of uveitis. However, the “privilege” does not offer protection from immune effects. Intraocular immune reactions are common, and several targeted immune pathway therapies have been developed for the treatment of uveitis (16, 17).
Eyelid contact allergic dermatitis as a result of ophthalmic drug administration is well established (ophthalmic preservatives, antibiotics, steroids, β-blockers [18⇓⇓–21]) indicating that surrounding ocular tissues are susceptible to the delayed hypersensitivity response (type IV) mediated by T lymphocytes. Irritant conjunctivitis is frequently confused with drug-induced conjunctivitis but represents a toxic insult to epithelial cells as opposed to a cell-mediated hypersensitivity response.
Considering APIs have demonstrated the ability to cause sensitization, it would stand to reason that leachables within ophthalmic products would carry an inherent risk as well. A guinea pig maximization test or murine local lymph node assay (LLNA) may not be directly relevant to the eye; however, if a positive reaction was obtained in either assay with a leachable compound, there may be sufficient weight of evidence to conclude the compound would have a high likelihood of stimulating an immune response leading to sensitization either within the eye or to the surrounding periorbital skin. The primary sensitization concern from the regulatory agency perspective is skin sensitization, and quantitative risk assessments related to contact allergies for skin sensitizers have been proposed (22⇓–24). The threshold for induction of sensitization in the existing skin sensitizer quantitative risk assessments relies upon identification of no-expected-sensitization-induction levels (NESILs) utilizing data from human repeated-insult patch test and correlation to LLNA resultant EC3 values, or the concentration expected to give a threefold stimulation of lymph node cell proliferation. A chemical can be assigned a potency classification based on their LLNA EC3 values (Table II). Collection of human data for the sensitization potential of an extractable or leachable would be considered excessive; however, human risk should be taken into consideration when assessing dosing frequency and duration for a product with potentially sensitizing extractables/leachables. A similar approach to what has been proposed for skin sensitizers and good scientific judgment is appropriate when assessing the potential for ocular drug-induced sensitization.
LLNA Potency Classification
Prediction performances of expert in silico structure–activity relationship (SAR) systems continue to improve; however, quantitative SAR cannot currently predict skin sensitization potential reliably (25, 26). Relative alkylation index models (also known as quantitative mechanistic models) may provide an approach to determination of sensitization potential based on the reactivity and hydrophobicity of molecules (27, 28).
Many ocular diseases have a genetic component (e.g., deoxyribonucleic acid [DNA] alterations) to their etiologies. These DNA alterations can cause adverse effects such as direct degeneration of tissue or upregulation of a growth factor that produces unwanted neovascularization (29). Additionally, environmental factors such as exposure to cigarette smoke and sunlight or a person’s diet can also impact individual susceptibility for a variety of degenerative ocular diseases.
Although systemic exposure following ocular drug administration is typically very low, alignment with established genotoxic impurity guidance when assessing E&L is warranted. Beyond the potential for local carcinogenic effects, topical ocular drugs often drain through the nasal lacrimal duct into the nasal cavity where systemic exposure is facilitated by either local absorption or swallowing.
Extractables/leachables provide no therapeutic benefit to the patient, and genotoxic risk would be unacceptable. If the chemical structure of the proposed leachable produces in silico structure–activity alerts, in vivo and in vitro genetic toxicity assays may need to be conducted in accordance with existing guidelines on genotoxic impurities. Testing can be performed on the drug product after accelerated storage conditions to maximize the presence of the leachable as long as the amount is greater than previously observed in nonclinical or clinical batches. Alternatively, genotoxicity assays can be performed with the purified leachable alone or spiked into the drug product.
Safety Qualification for Leachables in ODPs
Qualification of leachables for ODPs is similar to that of other drug types, although the toxicological endpoints of concern would be more focused on local toxicity rather than systemic effects. As for other product types, limits and/or qualification are usually established on a case-by-case basis using safety assessments in conjunction with quality manufacturing considerations.
If an extractable study is conducted, structure–activity relationship evaluations (e.g., expert-ruled and statistical quantitative structure–activity relationship (QSAR), per ICH M7) of the extractables for genotoxicity potential may be conducted as part of the early risk assessment. Additionally, a screening-level literature review could also be conducted to evaluate for any significant toxicities associated with the compound. This early toxicological assessment could serve to highlight extractables that may require specific monitoring later in the leachable study or if there are compounds of high concern identified, could result in the decision to replace the source of the unwanted leachables with an alternative material.
For qualification of an ophthalmic leachable, a literature evaluation should be conducted, focusing on the local effects including ocular toxicity, potential for carcinogenicity/genotoxicity, and sensitization. Systemic data may be used to support the qualification; however, it is not considered of primary concern.
Based on the structural alert information from the SAR assessment and the literature search, along with the potential for human exposure/duration of use, the toxicologist would perform a safety assessment and determine whether the literature provides adequate information to qualify the leachable or if additional testing is required. The safety assessment should evaluate local effects on the eye (i.e., ocular irritation/toxicity) on a concentration basis (parts per million); however, for evaluation of genotoxicity potential, it may be useful to also calculate the TDI to aid in the assessment of systemic risk. Any additional testing would be focused on areas where there were data gaps for the primary endpoints of concern (ocular irritation/toxicity, sensitization, genotoxicity).
Conclusion: Toxicological Considerations for Leachables in ODPs
Qualification of leachables for ODPs is similar to that of other drug types; however, the toxicological endpoints of concern would be focused on the potential for local toxicity rather than systemic effects. The primary toxicological endpoints that need to be considered for qualifying leachables for topical ophthalmic products include ocular irritation and toxicity; sensitization (skin), and genotoxicity. The safety assessment should evaluate local effects on the eye (i.e., ocular irritation/toxicity) on a concentration basis (parts per million); however, for evaluation of genotoxicity potential, it may be useful to also calculate the TDI to aid in the assessment of systemic risk. The need for additional testing is focused where there were data gaps for the primary endpoints of concern (ocular, sensitization, and genotoxicity).
Case Study on the Safety Qualification of Leachables in an ODP
A leachable study was conducted in parallel with a registration stability program for a novel topical ODP. The configuration for the leachable study consisted of the ODP in the primary packaging along with key secondary packaging components including the label, the product information insert, and the unit carton. Additionally, negative control samples, which consisted of the drug product stored in impermeable glass vials, were also included in the study to be evaluated if needed to differentiate potential leachables from direct formulation-related species.
This drug product is under development for two separate potential applications with differing dosing regimens: (i) four times daily (for a short-term indication for use in the affected eye(s) (≤ 14 days) and (ii) twice daily for a chronic indication to be used in both eyes.
In the leachable study, six peaks were detected and confirmed to be leachables based on comparative studies with the negative control (Table III). Two of these species were at or near the analytical limit of detection and could not be positively identified.
Summary of Leachables
For the other four identified leachables, the available analytical concentration data are summarized in Table III. Local effects are the primary parameter evaluated in terms of concentration on the eye (parts per million); however, it is useful to calculate the TDI to aid in the assessment of genotoxicity potential. The total daily exposure for each leachable and for each indication was calculated using the following formulas:

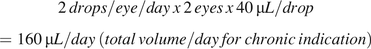
Based on the volume administered/day, the total daily ocular exposure for each leachable was calculated as:

Based on the low concentrations (≤1.5 ppm) and potential systemic exposure levels, it was deemed that no further work or safety assessments were required for the two unidentified chemicals (peaks # 5 and 6) and for peak #3. For the other three compounds, which were detected at concentrations in the 7–22 ppm concentration range and near or above the threshold of toxicological concern (1.5 µg/day) for total daily exposure (30), literature searches focused on the three primary toxicity endpoints for this product type were conducted. A high-level summary of the literature data is found in Table IV. The evaluation of each of the three compounds is described next.
Summary of Available Literature for Key Leachables
Antioxidant Compound
As only minimal, reversible ocular irritancy/toxicity was observed at very high concentrations (800-fold higher than product), it was deemed that no further ocular testing was required. As no data was available for genotoxicity and the TDI was above the TTC, SAR analysis was conducted. No SAR alerts for genotoxicity were detected. Based on these results, along with the overall dataset, no additional safety testing is required.
Plasticizer
For this material, favorable ocular toxicity data (at higher concentrations than detected in the product), as well as favorable genotoxicity and sensitization data were found in the literature. Based on the available data and the exposure of the plasticizer relative to the recommended safe level on California Proposition 65, no additional testing is required for this material.
Resin Intermediate
For the resin intermediate detected at 22 ppm in the drug product, there was minimal ocular data available at relevant concentrations. Therefore, further evaluation/testing of ocular irritation/toxicity may be warranted. The favorable genotoxicity and sensitization data available in the literature precludes any further testing in these areas.
The literature review and safety assessment of these materials along with any proposed testing strategies should be documented for potential inclusion in future regulatory submissions.
General Sample Evaluation (with Examples of Assessment Tables)
No-observed-adverse-effect-levels (NOAELs) from local tolerance, sensitization, and genotoxicity studies should be identified from published studies whenever possible. If insufficient information is available, additional safety parameters such as SAR assessments and acute and chronic systemic toxicity can contribute to the safety evaluation. If no information is available, structurally related compounds may be used on a case-by-case basis, as appropriate, to provide an assessment from similar compounds. See example sample assessment tables, Table V and Table VI.
Sample Assessment: Highest Concentrations Detected and Estimated Total Daily Doses
Example Tool to Aid in Assessment (Add or Delete Categories as Applicable). Complete for Each Leachable
Conclusion: Case Study
Literature data assessing local tolerance, sensitization, and genotoxicity should be evaluated as part of the safety assessment and to determine if additional testing is required. As part of the safety assessment of local effects, the total daily ocular exposure for each leachable should be calculated, considering the clinical dose regimen. In addition, the TDI of the drug should be calculated in support of a genotoxicity assessment. The literature review and safety assessment along with any proposed testing strategies should be documented for potential inclusion in future regulatory submissions.
Conflict of Interest Declaration
The authors declare that they have no competing interests.
Acknowledgment
The support of the Product Quality Research Institute and, specifically, the PQRI Working Group for Extractables & Leachables in Parenteral and Ophthalmic Drug Products (PODP), is gratefully acknowledged.
Footnotes
PDA PAPER DISCLAIMER: The following paper is a special contribution from the Product Quality Research Institute (PQRI) Leachables and Extractables Ophthalmic Sub Team. This article was internally reviewed by PQRI working group members and not peer-reviewed by the PDA Journal of Pharmaceutical Science and Technology. This paper is protected by copyright and unauthorized distribution or use is prohibited.
- © PDA, Inc. 2022
References
In This Issue




{kind=link}
Jump to section
- Article
- Abstract
- Background
- Importance of Assessing Leachables in ODPs
- Current Regulatory Approaches
- Specific Considerations for Managing Leachables in ODPs
- Potential Sources and Common Classes of Leachables for ODPs
- False Negatives Observed during Extractable Testing of Secondary Packaging Components and Leachable Testing of Drug Products for Which Secondary Packaging Components Are Critical
- Experimental Design Considerations: Extraction/Simulation Studies for ODPs
- Experimental Design Considerations: Leachable Studies for ODPs
- Analytical Evaluation Threshold
- Summary of Analytical Challenges for Extractables and Leachables in ODPs
- Toxicological Considerations for Leachables in ODPs
- Safety Qualification for Leachables in ODPs
- Conclusion: Toxicological Considerations for Leachables in ODPs
- Case Study on the Safety Qualification of Leachables in an ODP
- General Sample Evaluation (with Examples of Assessment Tables)
- Conclusion: Case Study
- Conflict of Interest Declaration
- Acknowledgment
- Footnotes
- References
- Figures & Data
- References
- Info & Metrics
Related Articles
Cited By...
- No citing articles found.