
Abstract
N-nitrosamines are chemical entities, some of which are considered to be possible human carcinogens, which can be found at trace levels in some types of foods, tobacco smoke, certain cosmetics, and certain types of rubber. N-nitrosamines are of regulatory concern as leachables in inhalation drug products, particularly metered dose inhalers, which incorporate rubber seals into their container closure systems. The United States Food and Drug Administration considers N-nitrosamines (along with polycyclic aromatic hydrocarbons and 2-mercaptobenzothiazole) to be “special case” leachables in inhalation drug products, meaning that there are no recognized safety or analytical thresholds and these compounds must therefore be identified and quantitated at the lowest practical level. This report presents the development of a quantitative analytical method for target volatile N-nitrosamines in a metered dose inhaler drug product, Atrovent® HFA. The method incorporates a target analyte recovery procedure from the drug product matrix with analysis by gas chromatography/thermal energy analysis detection. The capability of the method was investigated with respect to specificity, linearity/range, accuracy (linearity of recovery), precision (repeatability, intermediate precision), limits of quantitation, standard/sample stability, and system suitability. Sample analyses showed that Atrovent® HFA contains no target N-nitrosamines at the trace level of 1 ng/canister.
- Special-case leachables
- N-nitrosamine analysis
- Metered dose inhalers
- Gas chromatography/thermal energy analysis detection
- Inhalation drug products
Introduction
Metered dose inhalers (MDIs) are a particular class of inhalation drug product that contain an active pharmaceutical ingredient(s) and excipients, either in solution or suspension, in a propellant (e.g., chlorofluorocarbon or an alternate propellant). The drug product formulation is contained within a metal canister under pressure, and the unit dose is delivered via a dose metering valve. The valve can incorporate rubber sealing components, such as gaskets and seats, which contain organic chemical additives (e.g., antioxidants, vulcanization agents, etc.) at trace levels. These organic chemical additives, along with their degradation products and impurities, can potentially leach into the propellant-based formulation over its shelf life.
The Product Quality Research Institute (PQRI) Leachables and Extractables Working Group (made up of representatives from the pharmaceutical industry, academia, and regulatory authorities, and charged with solving fundamental technical problems to improve drug product quality and safety) released a recommendation document that includes safety and analytical thresholds for leachables in orally inhaled and nasal drug products (OINDPs, or inhalation drug products), such as MDIs. The document also provides best practice recommendations for the conduct of OINDP pharmaceutical development programs regarding leachables and extractables (1). These recommendations have also been published separately in the scientific literature (2, 3). The safety thresholds and best practice recommendations were developed to cover the diversity of organic chemical entities that can appear as leachables in inhalation drug products. The PQRI working group also recognized that certain potential leachables with special safety and historical concerns should be grouped for separate consideration, creating the so-called “special case” compounds and compound classes, including polycyclic aromatic hydrocarbons (PAHs), N-nitrosamines, and 2-mercapto benzothiazole (2-MBT), all of which must be evaluated by specific analytical techniques and technology-defined thresholds (1).
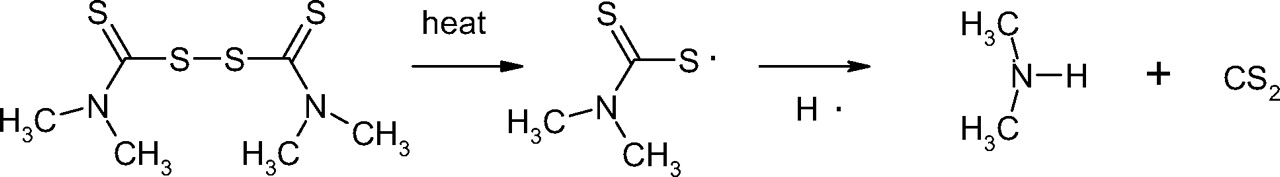
N-nitrosamines are a class of organic chemicals with basic molecular structure as shown in scheme I where R represents alkyl (e.g., methyl, ethyl, butyl) or other functionalities (e.g., piperidine, morpholine), some of which have been classified as possible human carcinogens (4). Volatile N-nitrosamines (see below) form when secondary amines react with certain “nitrosating agents” such as NO+, N2O3, and N2O4 (4). Volatile N-nitrosamines have been shown to occur at trace levels in water and wastewater (5–19), foods (20–32) and certain types of cooked meats in particular (32), tobacco smoke (33, 34), cosmetics (35, 36), rubber (4, 37–44), and certain pharmaceuticals (45, 46). Non-volatile and other more unusual N-nitrosamines have also been reported in pharmaceuticals (47, 48). Secondary amines can form in certain types of rubber from vulcanization curing agents, such as thiurams and thiocarbamates (37). For example, tetramethylthiuram disulfide can form N-nitrosodimethylamine (NDMA) as shown in scheme II.
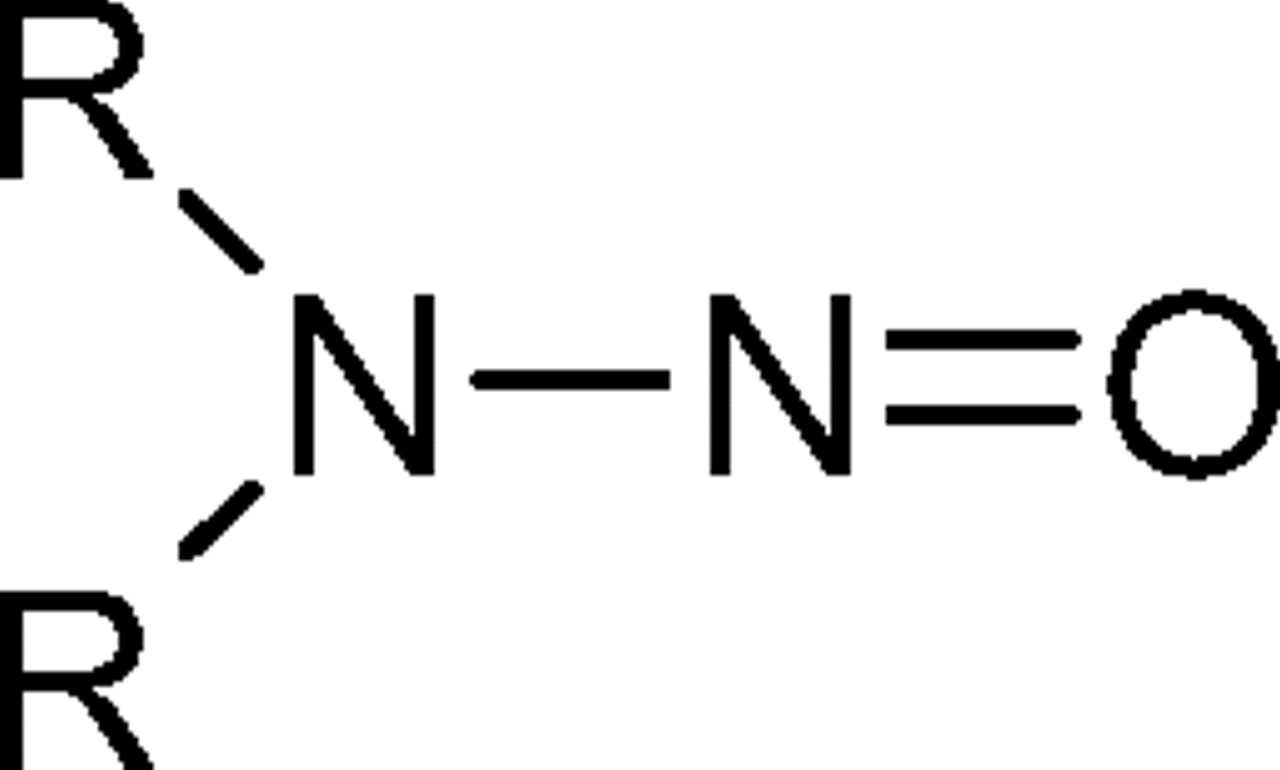
Chemical Structure for N-nitrosamines

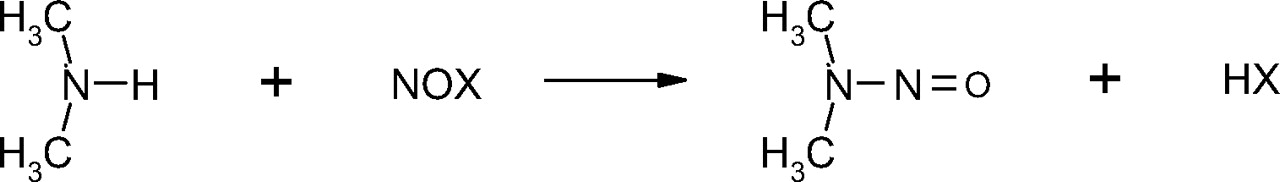
Scheme for the formation of N-nitrosodimethylamine (NDMA) from tetra-methylthiuram disulfide.
Volatile N-nitrosamines would then be considered as trace level impurities in the finished rubber matrix, with known rubber additives as precursors.
Concern regarding trace levels of volatile N-nitrosamines in rubber dates back to the discovery of N-nitrosamines in baby bottle nipples and pacifiers (38) and has led to the development of specific and highly sensitive analytical methods for target volatile N-nitrosamine analytes in rubber (38, 39), including an Association of Official Analytical Chemists (AOAC) official method (49). These analytical methods for target volatile N-nitrosamines in rubber employ the highly specific and sensitive GC/TEA system (Gas Chromatograph/Thermal Energy Analysis® detector) (50), which is based on the phenomenon of chemiluminescence. Volatile N-nitrosamines eluting from the gas chromatograph are pyrolyzed (500 °C) and form nitrosyl radicals. The nitrosyl radicals then react with ozone (under vacuum) and form an excited state of NO2. The NO2 relaxes to ground-state and emits photons in the red and infrared regions (chemiluminescence) which are detected by a spectrophotometer.
GC/TEA has also been used to analyze volatile N-nitrosamines in foods (22, 23, 29). Other techniques used for analyzing volatile N-nitrosamines in various matrices include gas chromatography/mass spectrometry (GC/MS) (7, 10–13, 15, 16, 18, 24), gas chromatography/nitrogen phosphorus detection (GC/NPD) (9, 12, 19, 20, 41, 42), gas chromatography/flame ionization detection, (GC/FID) (12), electrochemical detection (8), liquid chromatography/mass spectrometry (LC/MS) (5, 6, 14, 28), and high-performance liquid chromatography (HPLC) with fluorescence detection (26). Additional technical and regulatory information regarding N-nitrosamines and other “special case” compounds in inhalation drug products is available (51, 52).
This report describes the development of an analytical method for target volatile N-nitrosamines in an MDI drug product, Ipratropium Bromide Monohydrate (HFA-134a) Inhalation Aerosol 0.021 mg TTV, 10 mL (referred to as Atrovent® HFA). Atrovent® HFA contains the aforementioned active pharmaceutical ingredient in a solution of HFA-134a propellant and ethanol. The method is based on GC/TEA so that a direct extractables/leachables correlation (1) can be accomplished with valve component rubber analyzed by the AOAC official method (49). The analytical method involves recovering the target volatile N-nitrosamines from the drug product matrix, followed by separation and detection by GC/TEA. While the sample preparation procedures in the AOAC method for rubber are different from the drug product leachables method described in this report, the methods share the same target analytes and analytical technique that facilitates the extractables/leachables correlation.
The capability, and therefore “validatability,” of the final analytical method was assessed with respect to specificity, linearity and range, accuracy (linearity of recovery), precision (repeatability, intermediate precision), limit of quantitation (LOQ), standard and sample stability, and system suitability. The method was designed to detect target N-nitrosamines at the low nanogram/canister level that was required based on the known quantitation limits of the rubber method (approximately 10 ppb for an individual target N-nitrosamine). No quantifiable target N-nitrosamines were detected in rubber associated with the dose metering valves of this MDI drug product.
Experimental Design
Final Analytical Method
Target analytes for the analytical method are
-
N-nitrosodimethylamine (NDMA)
-
N-nitrosodiethylamine (NDEA)
-
N-nitrosodi-n-butylamine (NDBA)
-
N-nitrosopiperidine (NPIP)
-
N-nitrosopyrrolidine (NPYR)
-
N-nitrosomorpholine (NMOR)
These were used for method development and method capability assessment as a “custom nitrosamine mix,” with 100.0-μg/mL nominal concentration for each analyte (ethanol solution in amber ampules, AccuStandard). N-nitrosodiisopropylamine, 100.0 μg/mL nominal concentration (ethanol solution in amber ampules, AccuStandard) was used as an internal standard. Note that these volatile N-nitrosamines are also target analytes for rubber (49).
A sample for analysis includes 10 Atrovent® HFA canisters which are vented using a piercing device, allowing propellant evaporation. The canisters are then cut open using a tubing cutter, and the remaining liquid contents are transferred to a 250-mL separatory funnel. Methylene chloride (40 mL; Fisher Optima) is used to rinse the canisters in triplicate, and 100 mL of water (USP-grade) is then added to the separatory funnel. After liquid/liquid extraction, the organic layer is collected and is dried down to less than 1 mL under a stream of nitrogen in a conical test tube. The solution is transferred to a 1-mL volumetric flask containing the internal standard (N-nitrosodiisopropylamine) and filled to volume with ethanol rinses of the conical test tube.
The sample extract is then ready for GC/TEA analysis. N-nitrosodiisopropylamine was chosen as the internal standard because it is a chemical analog of the target compounds. Therefore, it will exhibit similar behavior in both the sample matrix and in the analysis as the target compounds without adding any chromatographic peak interference. Because relative response factors can be determined (target response/internal standard response), splitless injector effects can be dampened. The GC/TEA operating parameters are shown in Table I. An extraction matrix blank is prepared for each set of samples by adding 20 mL of USP ethanol, 40 mL of methylene chloride, and 100 mL of USP water to a 250-mL separatory funnel and then preparing the matrix blank for GC/TEA analysis as a sample.
GC-TEA Conditions for the Final Analytical Method
A typical sample analysis injection sequence includes two ethanol blanks, six replicates of a 100-ng/mL N-nitrosamine mixed standard, and duplicate injections of an extraction matrix blank followed by duplicate injections of sample extracts. Each set of six samples (12 injections) is bracketed by duplicate injections of the 100-ng/mL standard.
System suitability parameters for the analytical method include area ratio precision (<10% relative standard deviation, % RSD, for each analyte over six injections of a 100-ng/mL nitrosamine standard), chromatographic resolution between adjacent pairs of target nitrosamine peaks (R ≥ 1 for each adjacent analyte pair for one injection of a 100-ng/mL nitrosamine standard), and chromatographic peak tailing factor (TF < 1.8 for each analyte peak in one injection of a 100 ng/mL nitrosamine standard). Concentrations of individual target nitrosamines are calculated with relative responses factors derived from the 100-ng/mL N-nitrosamine standards over the entire sequence.
Method Capability Study
-
System Suitability: Injection precision, chromatographic resolution (R) of adjacent analyte peaks, and chromatographic tailing factors (TFs) of analyte peaks were evaluated from 10 replicate injections of a 100-ng/mL standard of target analytes and internal standard.
-
Linearity and Range: Linearity and range were evaluated with duplicate injections of mixed analyte standards and internal standard over the concentration range of 5.0 to 500 ng/mL (0.5–50 ng/canister) for NDMA and NDEA, and 12.5 ng/mL to 500 ng/mL (1.25–50 ng/canister) for the other four target analytes.
-
Limit of Quantitation (LOQ): LOQ values were estimated by extrapolating signal-to-noise ratio from analyses of the 25 ng/mL (2.5 ng/can) standard solution (10 injections). Noise refers to root-mean-square (RMS) noise which was estimated as 1/5 the peak-to-peak noise level of the baseline. Estimated LOQ values were confirmed with 10 injections of the 10-ng/mL (1.0-ng/canister) standard.
-
Accuracy (Linearity of Recovery): Vented canister samples and extraction blanks were spiked with target analytes at levels of 100 ng/mL, 200 ng/mL, and 300 ng/mL (10 ng/canister, 20 ng/canister, 30 ng/canister) using a 100-μL gas-tight syringe. The endogenous 100-ng/mL and 300-ng/mL spiked samples were prepared and extracted in triplicate, and the 200-ng/mL spiked samples were prepared in six replicates for use in the Precision (Repeatability) assessment.
-
Precision (Repeatability): Six spiked canister assays were accomplished as stated above at the 200-ng/mL level (20 ng/canister). Samples were analyzed in duplicate.
-
Intermediate Precision: Intermediate precision was assessed between two analysts, each of whom accomplished duplicate preparations of 500-ng/mL spiked samples along with an extraction blank. In addition, both analysts shared the same column and the same GC/TEA instrument.
-
Specificity: The extraction blank and spiked samples (200-ng/mL N-nitrosamine mix) from the accuracy experiment, and a freshly prepared 100-ng/mL N-nitrosamine mix, were subjected to at least 12 h of exposure to UV light, followed by at least 12 h of exposure to white light. Extraction blank, spiked samples, and standards were reanalyzed for remaining nitrosamines.
-
Standard and Sample Solution Stability: Standard stability was determined by analyzing, in duplicate, a 500-ng/mL standard stored in an amber volumetric flask on the countertop after 24, 48, and 72 h. Sample stability was determined by analyzing, in duplicate, a 500-ng/mL (50 ng/canister) spiked sample stored in an amber autosampler vial on the countertop after 24, 48, and 72 h.
Drug Product Samples for Analysis
Atrovent® HFA drug product canisters used for method development, method capability assessment, and sample analysis studies were taken from either development batches or New Drug Application (NDA) primary stability batches, all of which incorporated the final container closure system configuration and drug product formulation.
Results and Discussion
Method Development
The method development process began with the GC/TEA conditions suggested by the AOAC official method for analysis of target N-nitrosamines in rubber (49), with a target LOQ of 1 ng/canister of Atrovent® HFA based on analyses of valve rubber components determined by the AOAC method, which had been validated for the particular rubber. A packed column-based GC/TEA method has the advantages of increased sample loading capability and ruggedness when subjected to harsh sample matrices. It was determined that the initial chromatography conditions, based on the packed column AOAC method, were inadequate due to a peak in drug product samples that partially coeluted with NDMA.
Various packed and capillary column configurations with optimized GC temperature parameters were evaluated (see Table II) before the final analytical method conditions used in Table I had been established. The greater separating capability of the capillary column in the chosen final chromatographic configuration compensated for reduced analyte loading capacity. It was also determined during the method development process that a guard column (IP Deact 5 m × 0.53 mm; Restek) was required in order to protect the analytical capillary column from excessive contamination and damage from the sample extract matrix. Furthermore, the recovery solvent, analyte recovery procedure, internal standard identity and concentration, TEA parameters, and GC injector temperature were also investigated and optimized during the method development process. The conditions and parameters chosen for the final analytical method gave the best compromise between analyte recovery, sample quality, and simplicity.
Summary of Analytical Method Development (Chromatographic Configurations)
Method Capability
-
System Suitability: Injection precision (assessed as % RSD) ranged from 0.75% to 5.70% for target compound peak area responses, and 4.94% to 5.67% for relative response factors (area target/area internal standard). Chromatographic resolutions of peak pairs ranged from 3.05 (% RSD 0.52) for NMOR/NPYR to 33.64 (% RSD 0.71) for NDMA/NDEA. Chromatographic TFs ranged from 1.18 (% RSD 0.90) for NDEA to 1.46 (% RSD 1.14) for NDMA. Final system suitability criteria were chosen for the final analytical method based on these results.
-
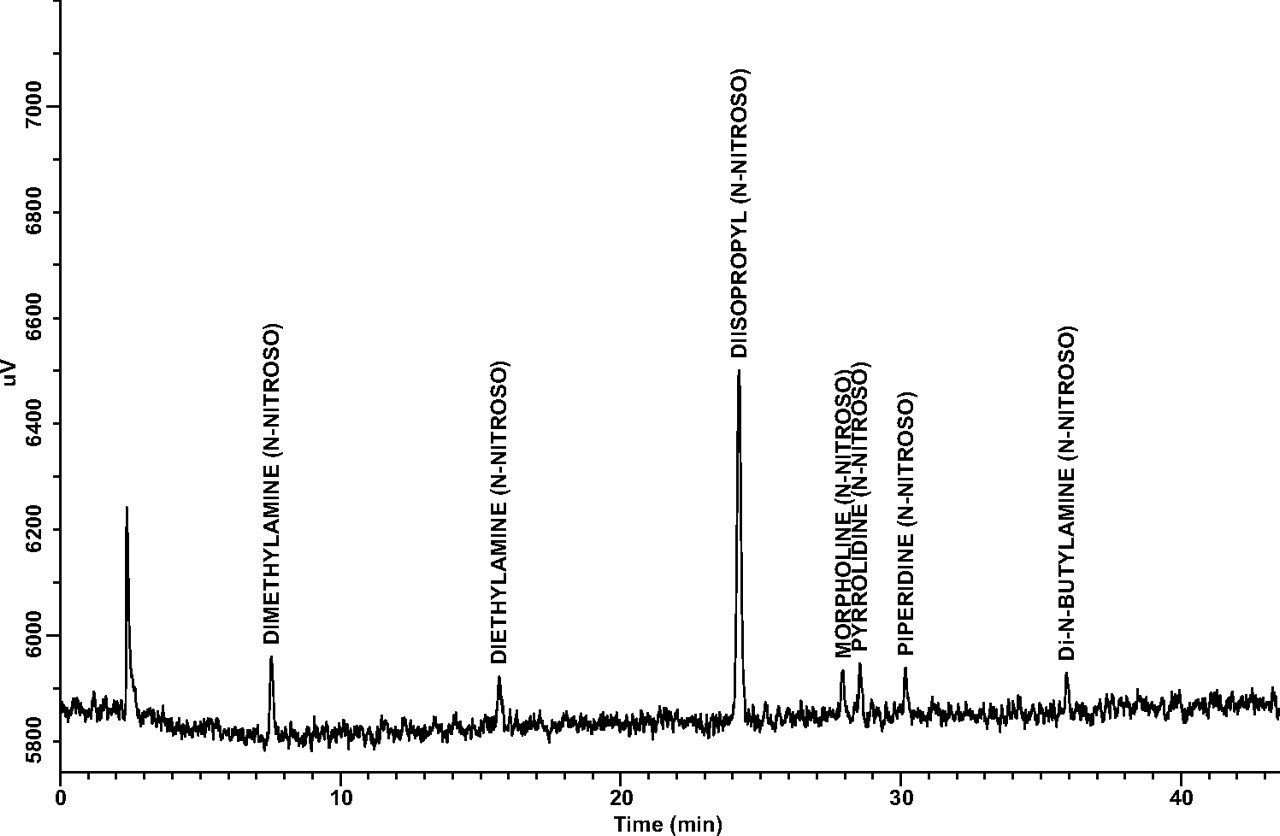
Linearity and Range: Linearity and range results for each target analyte are presented in Table III. Note that correlation coefficients (r2) were greater than 0.99 for all target analytes over the established concentration ranges, and that the zero point (origin) is within the 95% confidence interval of each y-intercept. A representative GC/TEA chromatogram of the 100-ng/mL N-nitrosamine target compound standard is shown in Figure 1.
-
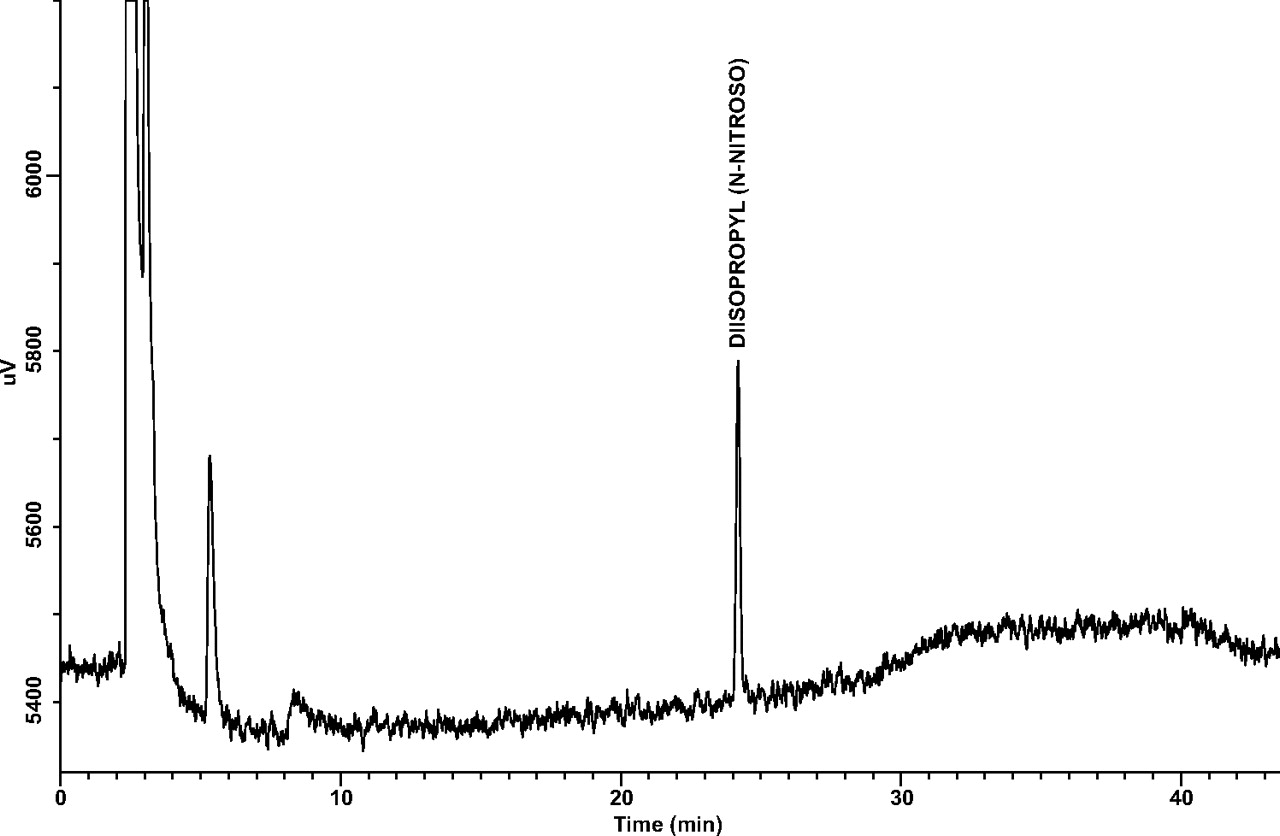
Limit of Quantitation (LOQ): LOQ values for each target N-nitrosamine estimated from the 25-ng/mL and 10-ng/mL standards are shown in Table IV. Note that all estimated LOQ values are at the target LOQ level of 1 ng/canister. A representative GC/TEA chromatogram for a 10-ng/mL (1-ng/canister) standard is shown in Figure 2.
-
Accuracy (Linearity of Recovery): Accuracy, as assessed by spiked analyte recovery at the three different spiking levels, is summarized in Table V. Representative GC/TEA chromatograms of an endogenous sample preparation and a 100-ng/mL spiked sample are shown in Figures 3 and 4. Note that certain analytes, in particular NDMA and NMOR, exhibited lower recoveries compared to the other four compounds. However, the well correlated linearity of recovery (amount recovered plotted versus amount spiked) behaviors shown by all target analytes compensate for these lower recoveries. Note that r2 values for each target analyte are ≥0.99. This suggests that measured recoveries could be used as correction factors for target analyte concentrations determined by the final analytical method.
-
Precision (Repeatability): Repeatability is also shown in Table V, based on the results from the 200-ng/mL spiked sample (six replicates). Note that % RSDs for all target analytes are <8%, again suggesting that measured recoveries could be used for analyte concentration correction.
-
Intermediate Precision: Intermediate precision results are shown in Table VI. Note that the percent difference values between analysts 1 and 2 were <20% except for NDMA. This suggests that careful and contemporaneous analyte recovery assessments through the analysis of spiked samples should be accomplished and perhaps included in the final analytical method.
-
Specificity: There were no target nitrosamines present in the standard, spiked sample, or extraction blank after exposure to at least 12 h of UV light and at least 12 h of white light (Figure 5). Clearly, chromatographic resolution is sufficient to separate target analytes from any potential chemical interferences. Furthermore, there were no interferences of the analytes from the drug product formulation.
This experiment also suggests a process for determining if any unknown peaks (i.e., peaks not corresponding to any target analyte) in a GC/TEA chromatogram are in fact N-nitrosamines. N-nitrosamines in solution are clearly unstable when exposed to light; therefore any unknown GC/TEA peak remaining after light exposure, such as in this study, could be concluded not to be an N-nitrosamine. An example of such an unknown GC/TEA peak is clearly shown in Figure 5 at a retention time of between 5 and 6 min.
-
Standard and Sample Solution Stability: Results for standard and sample stability are shown in Tables VII and VIII. The 100-ng/mL standard solution was deemed stable for up to 72 h (Table VII). The variability over the 72 h ranged from 99.6 to 110.7%. The 500-ng/mL spiked sample was also deemed stable for up to 72 h with a variability of 107.4 to 114.6%. The slightly higher variability for the spiked sample was deemed insignificant for the purposes of this study.
Linearity Results for Target N-nitrosamines. Nominal Concentration Range is 0.50 ng/Canister to 50.0 ng/Canister for N-nitrosodimethyl- and N-nitrosodiethylamines, 1.25 ng/Canister to 50.0 ng/Canister for All Others

A representative GC/TEA chromatogram of the 100 ng/mL N-nitrosamine target compound standard.
Limit of Quantitation (LOQ) Estimated from 10 Replicate Injections of 25 ng/mL and 10 ng/mL Standards which are Equivalent to 2.5 ng/Canister and 1.0 ng/Canister
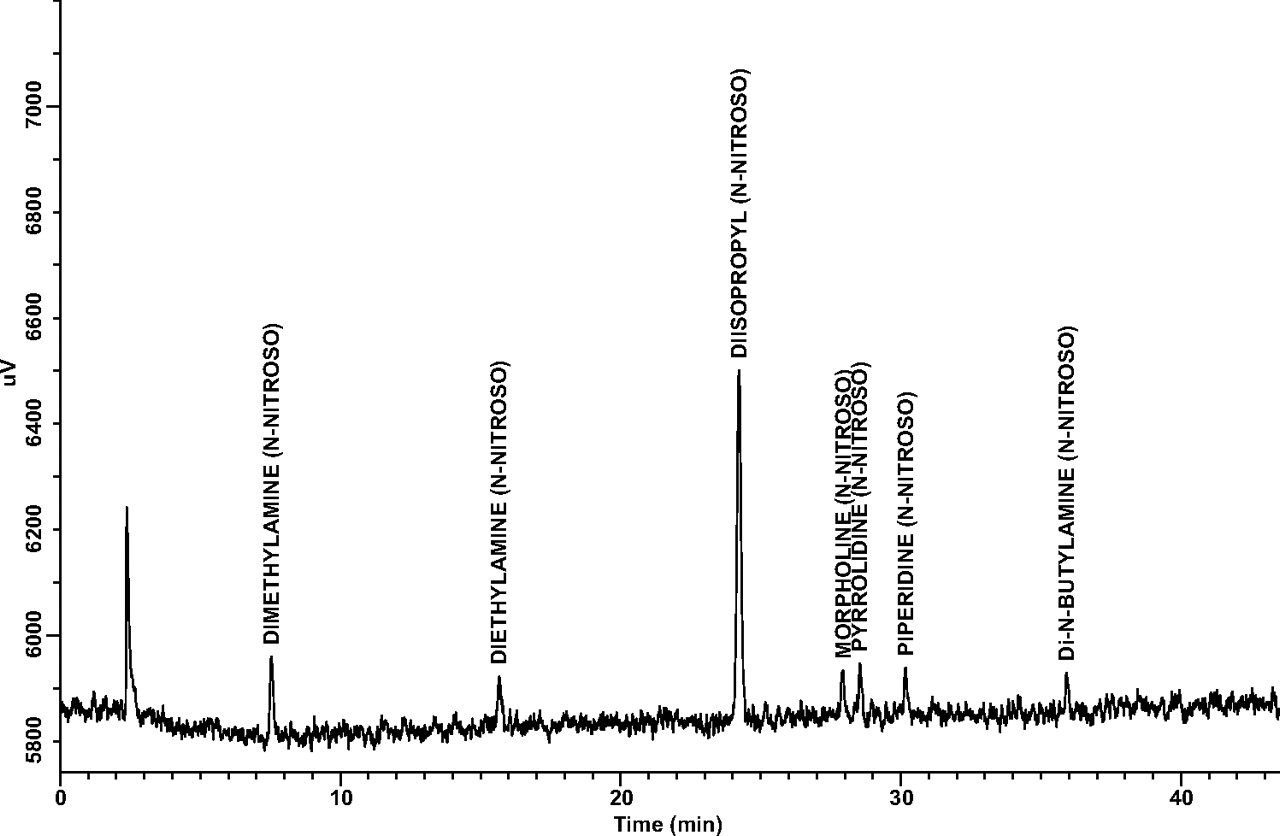
A representative GC/TEA chromatogram of an LOQ confirmation standard at 10 ng/mL (1.0 ng/canister).
Accuracy (Recovery). Linearity of Recovery Data for Spiked N-nitrosamines: Endogenous, 100, and 300 ng/mL Levels were Performed in Triplicate, while the 200 ng/mL Level was Performed with Six Replicates (Repeatability). N-nitrosodiisopropylamine (Internal Standard) was Spiked at 100 ng/mL in Each Sample

A representative GC/TEA chromatogram of an endogenous sample preparation.
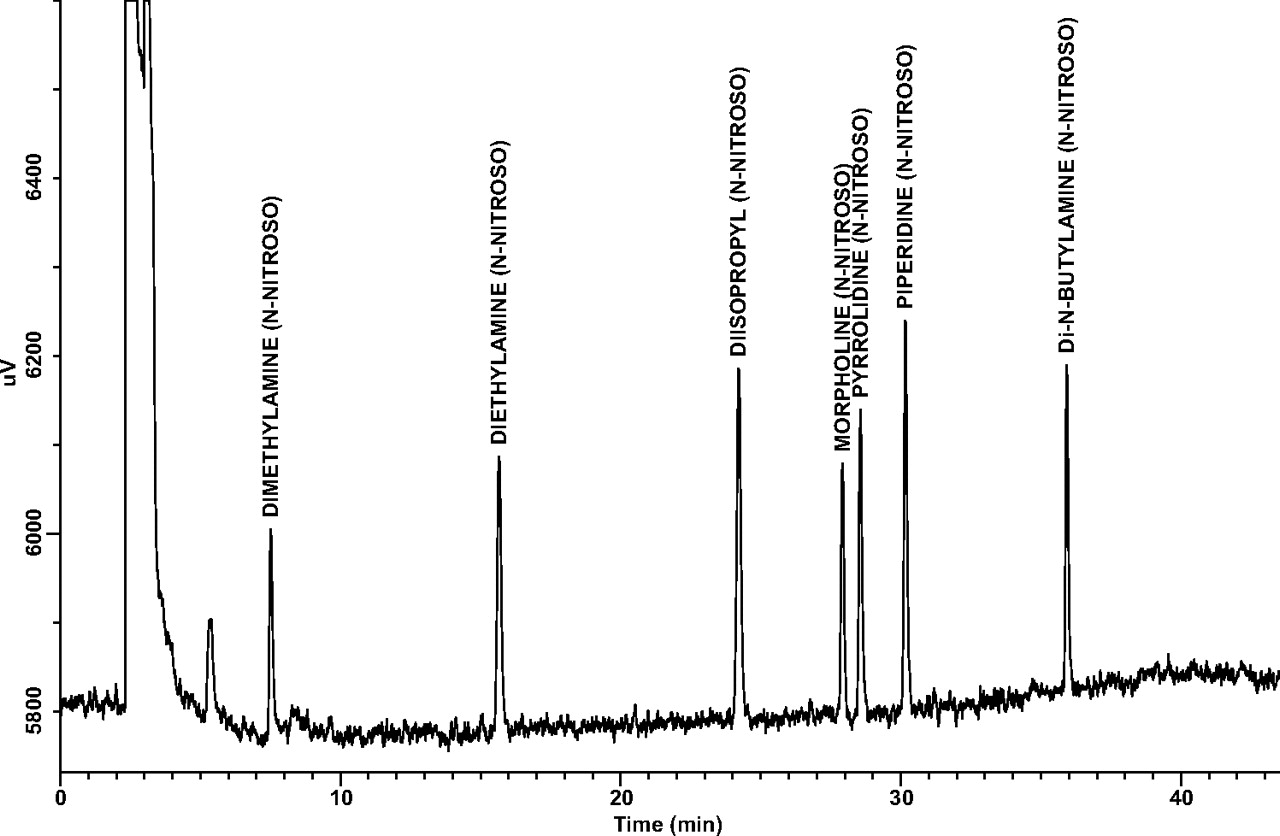
A representative GC/TEA chromatogram for a 100 ng/mL (10 ng/canister) spiked sample.
Intermediate Precision Results for the Nominal 500 ng/mL Spiked Samples

Representative GC/TEA chromatograms of a 200 ng/mL target N-nitrosamine spiked sample before (top) and after (bottom) exposure to at least 12 h of UV light, followed by at least 12 h of white light.
Standard Stability Results. Stock Stored in Amber Glassware on the Benchtop
500 ng/mL Spiked Sample Stability Results. Sample Stored in Amber Autosampler Vial on the Benchtop
Sample Analysis
Three stability batches of Atrovent® HFA (stored for 18 months at 30 °C/70% RH, inverted) were analyzed in duplicate for target N-nitrosamines using the developed analytical method. In addition, spiked drug product canisters (spiked at 3 and 10 ng/canister) from one of the stability batches (canisters remaining in quarantine from the 12 month stability time-point, CRT/ambient quarantine storage conditions) were also analyzed. No target N-nitrosamines were detected in any of the 18 month stability samples (inverted orientation), whereas all target N-nitrosamines were detected in spiked samples. A representative GC/TEA chromatogram of a 3 ng/canister spiked sample is shown in Figure 6.

Representative GC/TEA chromatogram of a 3 ng/canister spiked sample.
Conclusions
A highly sensitive and specific analytical method has been developed for target volatile N-nitrosamines in Atrovent® HFA MDI drug product. Analysis of drug product samples from accelerated stability storage demonstrated that no target volatile N-nitrosamines were present. This result confirmed the hypothesis that no nitrosamines are present in the drug product. This hypothesis was based on analyses of rubber components from the dose metering valve, using a fully validated version of the AOAC rubber method (49), which revealed no detectable target volatile N-nitrosamines.
Although the analytical method was determined to be highly sensitive, specific, and suitable to demonstrate the absence of target N-nitrosamines from this MDI drug product, recovery values for certain target analytes were found to be less than desirable. However, excellent linearity of recovery, repeatability, and intermediate precision results suggest that measured recovery values could be used to correct analyte concentrations determined by the method. Analyte recoveries should be assessed with spiked samples, and measured contemporaneously with sample analyses. With appropriate modifications as suggested above, the analytical method is considered to be “validatable” as a quantitative impurity method.
Acknowledgements
The authors wish to acknowledge their respective organizations, Boehringer Ingelheim Pharmaceuticals, Inc. and Catalent Pharma Solutions (formerly Cardinal Health), for their support.
Footnotes
- © PDA, Inc. 2009



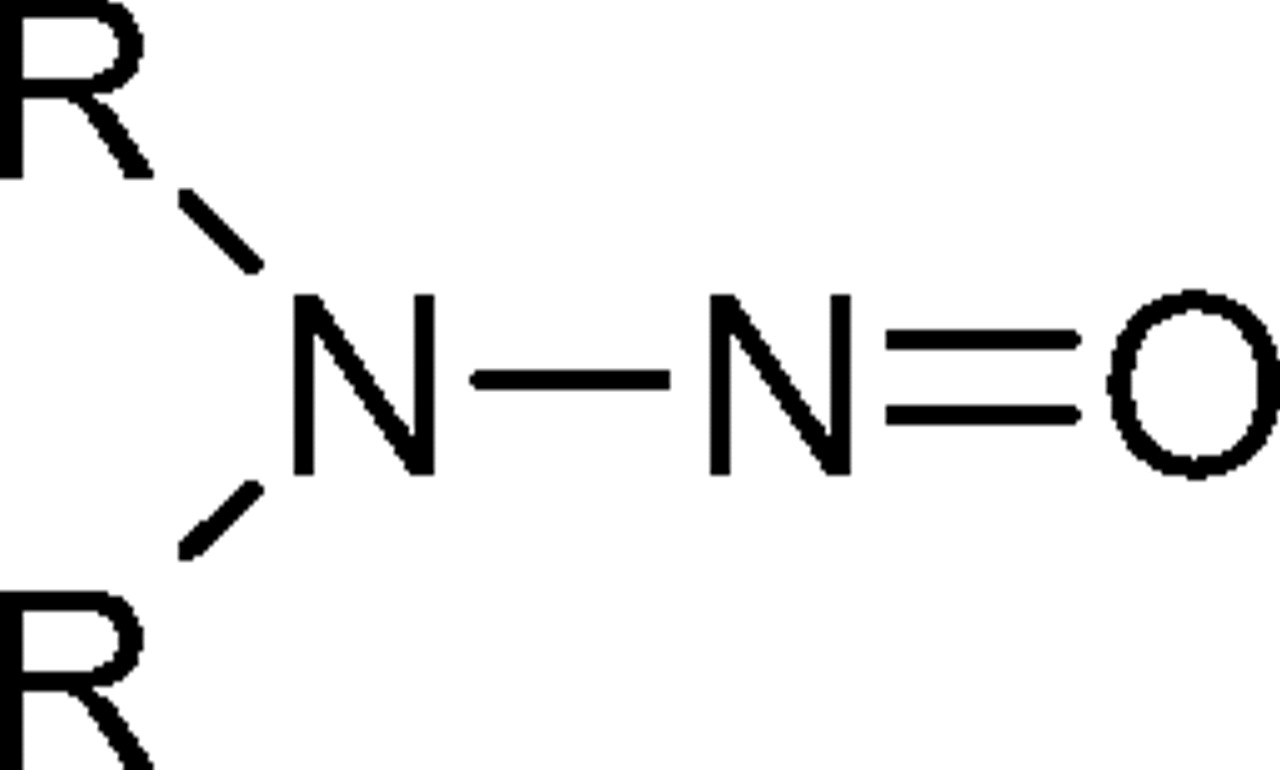{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}