
Abstract
Notable progress has been made in methods that encourage the use of polymerase chain reaction (PCR) as a rapid and accurate tool in microbiological testing of pharmaceuticals. In this study, the detection of the four main specified microorganisms according to the pharmacopeial recommendations, Salmonella spp, Escherichia coli, Pseudomonas aeruginosa, and Staphylococcus aureus, was optimized in different pharmaceutical dosage forms and raw materials. Uniplex PCR was performed for the detection of each microorganism individually targeting the conserved region in each bacterial genome. Further optimizations were done to perform duplex and multiplex PCR assays considering relative concentrations of competitor primers used in the reaction. The uniplex PCR amplicons were successfully sequenced, confirming the conservation of used primers. Other validation parameters such as specificity, sensitivity, and robustness were examined closely. The method provides a high-throughput screening method to test different pharmaceutical preparations for specified microorganisms for the detection of microbiological contamination.
LAY ABSTRACT: Strict regulations govern the production of pharmaceutical products whether they are sterile or nonsterile. Certain official tests are carried out in microbiology testing laboratory in any pharmaceutical production facility to ensure the pharmaceuticals microbiological quality according to the standard pharmacopeial recommendations. Nonsterile products must be free of specified microorganisms that are used as a check for their quality. Topical preparations must be free of Pseudomonas aeruginosa and Staphylococcus aureus, and oral preparations must be free of Salmonella spp and Escherichia coli. Conventional microbiological methods are time-consuming, labor-intensive, and require long incubation times, resulting in delaying the release of the products. In this study, we tested and validated a polymerase chain reaction identification approach to detect indicator bacteria in pharmaceutical preparations. The method depends on amplification of certain conserved genes located in the four specified bacteria. The method is optimized to be carried out individually or collectively to detect all indicator bacteria in a single reaction in different forms of pharmaceutical products.
Introduction
The microbial contamination of pharmaceutical raw materials and finished products represents potential hazards to consumers and usually leads to serious health problems. Several previously reported incidents were due to contaminants being present in raw materials and finished products, such as tetanus caused by contaminated talcum powder (1), Salmonella infections caused by contaminated thyrodine tablets (2), coloring agent (caramine) of capsules (3), and pencreatine powder (4). Other cases included Pseudomonads-contaminated eye cosmetics resulting in ocular ulcers (5), skin infections caused by Mycobacterium chelonae in gentian vilot (6), and, recently, septicaemia and conjunctivitis caused by Serratia marcescens–contaminated baby shampoo (7). Product recalls as a result of microbial contamination have been regarded as an issue resulting in huge financial loss to the manufacturing companies and the drug industry (8, 9). Whether the raw materials are active ingredients or excepients, those of natural sources are subjected to higher contamination levels than others of synthetic or mineral sources because of their characteristics and the series of manipulations they undergo. Excepients are mostly affected among the ingredients because the microorganisms use them as a substrate of their growth (10).
The USP determined the microbiological quality requirements of nonsterile pharmaceuticals (11). The microbial limit tests require the evaluation of given pharmaceutical samples quantitatively by determination of total microbial count and qualitatively by ensuring the absence of specified microorganisms that are health-threatening to consumers. Four indicator bacteria must be absent: Escherichia coli, Salmonella spp, Staphylococcus aureus, and Pseudomonas aeruginosa. Considering the route of administration and potential hazard of microorganisms to users, USP limits applied to oral preparations necessitate the absence of Escherichia coli and Salmonella spp, while those applied to topicals necessitate absence of Staphylococcus aureus and Pseudomonas aeruginosa as outlined in USP 34 〈1111〉. The microbial contamination of nonsterile pharmaceuticals may be attributed to contaminated raw materials, water, manufacturing processes, personnel, packaging materials, and storage conditions.
The use of rapid technologies for microbiological assessment in the pharmaceutical industry provides accurate, sensitive, robust, and fast evaluation of pharmaceutical preparations, allowing optimization of pharmaceutical process control and avoiding manufacturing losses and product recalls. Adenosine triphosphate (ATP) biolumensence, impedence, direct viable count, and flow cytometry determine the total microbial biomass in pharmaceutical sample, while immunoassay and polymerase chain reaction (PCR) detect the presence or absence of specific microbial species. PCR, which depends mainly on amplification of specific genetic sequence, has been used successfully as a rapid method of microbial detection in many fields, clinical samples (12⇓⇓–15), food products (16⇓⇓⇓–20), water monitoring (21⇓⇓⇓–25), and environmental samples (26, 27). During the last decade, several studies and experiments were carried out by different laboratory groups over the world for the optimization of these techniques and encouraging their use as a routine tool in the pharmaceutical industry (28⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓–39).
The aim of the present work is to optimize the use of PCR capable of detecting the four major microbial contaminants specified by the USP and to validate the application of these methods in the microbiological tests performed on nonsterile pharmaceutical products produced by The Nile Company for Pharmaceuticals and Chemical Industries, one of the major pharmaceutical companies in Egypt.
Materials and Methods
Bacterial Strains
All microorganisms used in the experiments were derived from the American Type Culture Collection (ATCC): Salmonella enterica serovar typhimurium ATCC 14028, Escherichia coli ATCC 8739, Pseudomonas aeruginosa ATCC 9027, and Staphylococcus aureus ATCC 6538. Cultures were prepared from KWIK-STICK PLUS Product, (Microbiologics, St. Cloud, MN, USA) according to the manufacturer's protocol. Tested microorganisms were harvested in tryptic soy broth (TSB) at 35 °C for 18 h and adjusted photometrically by using a PhoenixSpec Nephelometer (Becton Drive, NJ, USA) to reach cell density equivalent to approximately 0.5 McFarland standard (108 CFU/mL), then serially diluted to reach 100 (1 CFU) per gram or milliliter of sample. Other strains and species used for specificity: Escherichia coli ATCC 25922, Pseudomonas aeruginosa ATCC 27853, Pseudomonas putida ATCC 31483, Staphylococcus aureus ATCC 25923, Staphylococcus aureus ATCC 12600, Staphylococcus epidermidis ATCC 12228, Streptococcus pyogenes ATCC 19615, Enterococcus faecalis ATCC 29212, Bacillus subtilis ATCC 6633, Enterobacter aerogenes ATCC 13048, Klebsiella pneumonia ATCC 13883, and Salmonella enterica serovar typhimurium ATCC 13311.
Standard Sample Enrichment
Raw materials such as gum, maize starch, magnesium stearate, mannitol, pancreatin powder, and finished products of different composition such as syrups, creams, ointments, and gels were artificially inoculated. The samples of pharmaceutical raw materials and finished products were aseptically added to TSB for raw materials, or to TSB containing 0.5% lecithin and 4% Tween 20 for finished products, with dilution factor 1:10. After mixing, each of the enrichment broth was inoculated with mixed culture of Salmonella typhimurium ATCC 14028, Escherichia coli ATCC 8739, Pseudomonas aeroginosa ATCC 9027, and Staphylococcus aureus ATCC 6538, adjusted photometrically and by serial dilution to reach 102 CFU/mL, followed by incubation at 35 °C for 18 h. The dilution factor was modified to 1:100 or 1:1000 according to each product to achieve the recovery of inoculated microorganisms according to methodologies mentioned in the USP. After incubation, TSB enrichment broths were aseptically subcultured on several selective or differential media according to USP 34 〈62〉.
Genomic DNA Extraction
The mild lysis method, performed by Jimenez et al. (29), was applied with some modifications. After 18 h enrichment, a 100 μL aliquot of each broth was added to 200 μL of lysis buffer containing Tris (10 mM), ethylenediaminetetraacetic acid (EDTA) (1 mM), 3 μL of 20 mg/mL proteinase K solution, and 0.5% Tween 20 at pH 8.0. Samples were incubated for 20 min at 35 °C for cell lysis and cellular proteins degradation, followed by incubation for 10 min at 95 °C for lysis completion and proteinase K degradation. Another extraction method (other than the mild lysis) was done using the commercial kit GeneJet Genomic DNA Purification Kit (ThermoScientific, Rockford, IL, USA) according to the manufacturer's protocol; the use of such kit provided purification of extracted DNA, which was important for duplex and multiplex PCR and further sequencing for PCR products.
Primer Sequences and PCR
The target genes and the primers sequences are listed in Table I. Primers sequences originally from Karanam et al. (37) were checked for specificity and analyzed by National Center for Biotechnology Information/Basic Local Alignment Search Tool (NCBI/BLAST). The sequence for some primers was modified after REFSEQ analysis to achieve maximum specificity, and they were structurally analyzed for hairpin, self, and heterodimer formations by an oligonucleotide analyzer. Primers were synthesized by eurofins mwg operon (Ebersberg, Germany).
Primers and Product Sizes for PCR
PCR was performed in 50 μL reaction volume in reaction buffer including 2 mM MgCl2, 200 μM deoxyribonucleotide triphosphates dNTPs, 10 pmol of each primer, and 1.25 unit of DNA Taq polymerase. PCR was carried out using a PTC-200 thermocycler (MJ Research, Waltham, MA, USA) with initial denaturation at 95 °C for 5 min, followed by 25 cycles of denaturation at 95 °C for 30 s, annealing at 53.3 °C for 30 s, and extension at 72 °C for 1 min, followed by final extension at 72 °C for 7 min. Further optimizations were done to achieve duplex and multiplex PCR assays by changing the primers concentrations as follows: duplex PCR for detection of S. aureus and P. aeruginosa in topical preparations required using primers of concentrations 10 pmol and 7 pmol, respectively; for the detection of E. coli and Salmonella spp in oral pharmaceutical preparations, primers of concentrations of 5 pmol and 10 pmol were used, respectively; and for multiplex PCR targeting the four microorganisms, the primers concentrations were 5 pmol for S. aureus and E. coli, 3 pmole for P. aeruginosa, and 7 pmol for Salmonella spp. Ten microliters of each PCR amplicon were analyzed by gel electrophoresis in 1.2% agarose gel (Bioron GmbH, Ludwigshafen, Germany) stained with ethidium bromide (10 mg/mL), (Bioshop, Burlington, ON, Canada) using tris acetate EDTA TAE buffer and with electrical current 80 V. Bands were visualized under UV transilluminator (UVP, Upland, CA, USA) and photographed using Digi Doc gel documentation system (Bio-Rad, Hercules, CA, USA).
Sequence Analysis of PCR Amplicons
PCR products were purified by using QIAquick PCR DNA Purification Kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol and sequenced using ABI PRISM BigDye terminator cycle sequence system (Applied Biosystems, Carlsbad, CA, USA). DNA sequences obtained were analyzed with Sequencher software (Genecodes, Inc., Ann Arbor, MI, USA). Sequences were compared to the ones available at the gene bank.
Results
Detection of Pharmaceutical Contaminants by Conventional Methods
The samples of pharmaceutical raw materials and finished products were artificially inoculated with mixed cultures of four indicator bacteria: E. coli, S. aureus, P. aeruginosa, and Salmonella in TSB and incubated at 35 °C for 18 h. The recovery of inoculated microorganisms was validated by streaking the inoculated broths on selective/differential media. E. coli was identified as brick red colonies on MacConkey agar, S. aureus was identified as yellow colonies with yellow zones on mannitol salt agar, P. aeruginosa was identified as greenish fluorescent colonies on cetrimide agar, and Salmonella identified as red colonies with or without back centers on xylose lysine deoxycholate agar.
Detection of Pharmaceutical Contaminants by PCR
PCR was applied for detection of four bacterial contaminants of pharmaceuticals. The primers targeted the following genes: 16S rRNA for E. coli with an expected size 559 bp, mRNA nuclease for S. aureus with an expected size 461 bp, invA gene for Salmonella spp with an expected size 275 bp, and oprL gene in Pseudomonas aeruginosa with an expected size 709 bp.
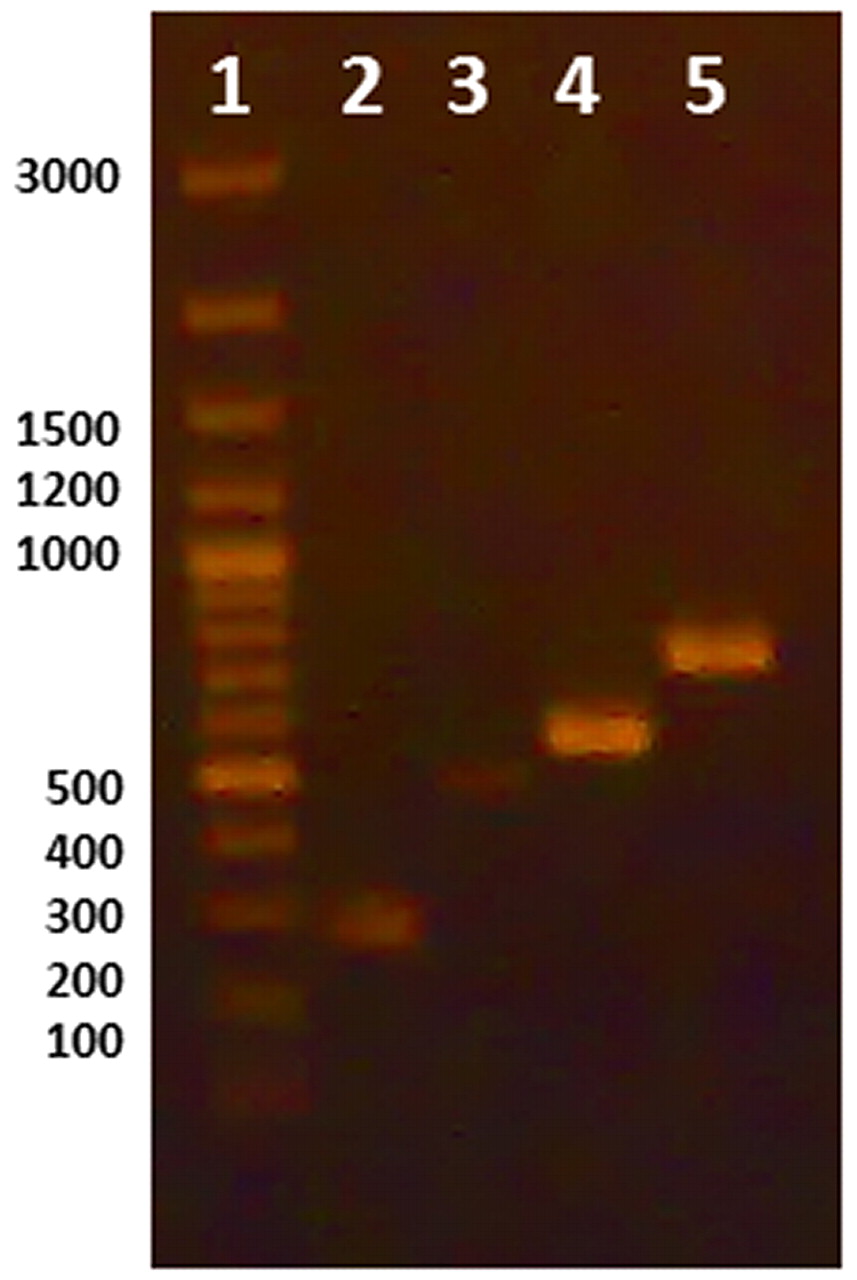
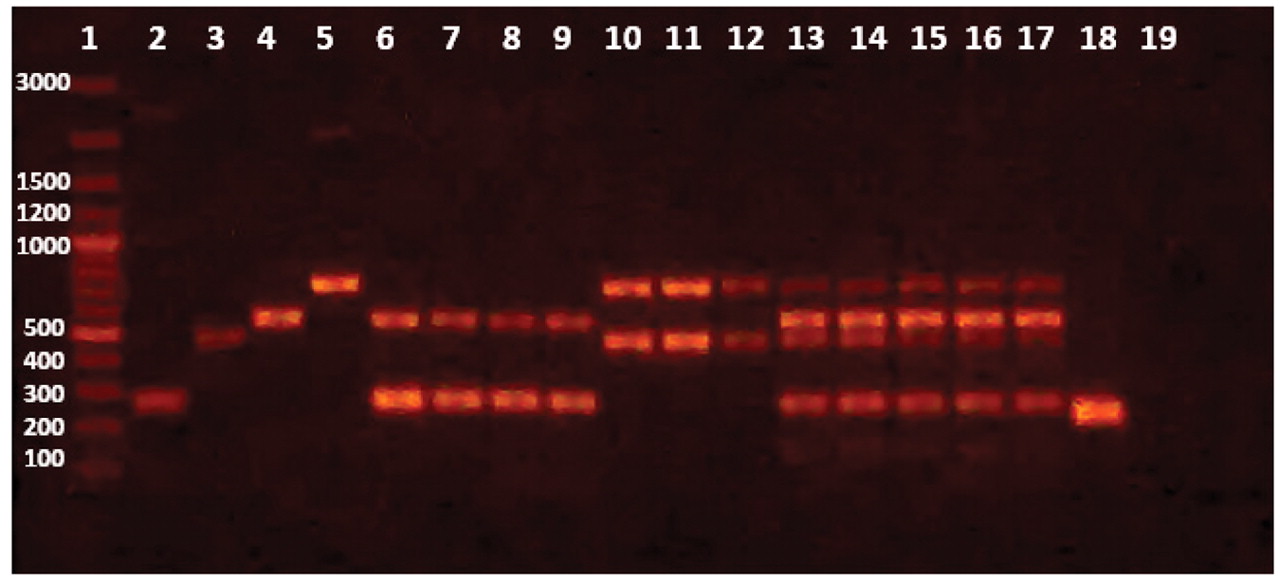
Initially, uniplex PCR was performed for detection of each microorganism independently. Single sharp distinct bands were observed for each corresponding microorganism at 2 mM MgCl2 and using 10 pmole of used primer with an annealing temperature at 53.3 °C (Figure 1). Further optimizations were done to achieve duplex PCR for detection of S. aureus and P. aeruginosa in topical preparations, E. coli and Salmonella in oral preparations, and all four microorganisms in one reaction by varying of concentrations of primers used for each microorganism. Figure 2 illustrates the PCR bands with uniplex, duplex, and multiplex PCR assays applied to different products from different categories of dosage forms and raw materials.

Uniplex PCR using DNA extracted by lysis buffer. Lane 1: Marker, Lane 2: Salmonella, Lane 3: S. aureus, Lane 4: E. coli, Lane 5: Pseudomonas aeruginosa.

Uniplex, Duplex and Multiplex PCR reactions applied to different nonsterile pharmaceutical products produced at The Nile Pharmaceutical Company. Band sizes are indicated next to the marker in base pairs. Lane 1: marker, Lanes 2–5: Uniplex PCR; 2: Salmonella, 3: Staphylococcus aureus, 4: E. coli and 5: Pseudomonas aeruginosa. Lanes 6–9: duplex PCR (Salmonella and E. coli) in four different syrups; 6–7: cough syrups, 8: anti-inflammatory, and 9: dietary supplement. Lanes 10–12: duplex PCR (Staph and Pseudomonas) in three topical preparations; 10: ointment, 11: gel, and 12: cream. Lanes 13–17: multiplex PCR in raw materials; 13: maize starch, 14: Mg stearate, 15: manitol, 16: pancreatin, and 17: gum. Lane 18: positive control (universal primer), Lane 19: negative control (no DNA).
Specificity, Sensitivity and Robustness of PCR
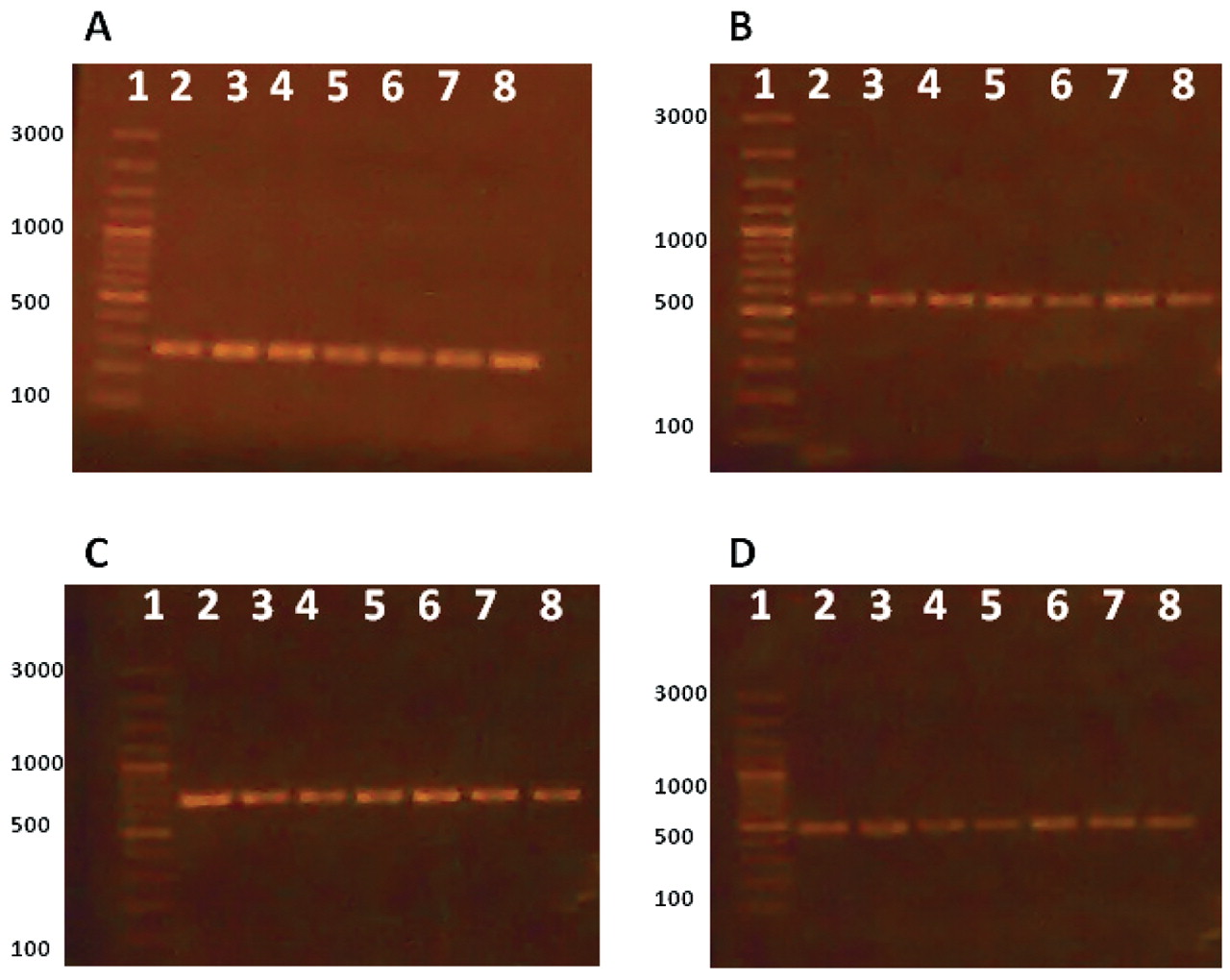
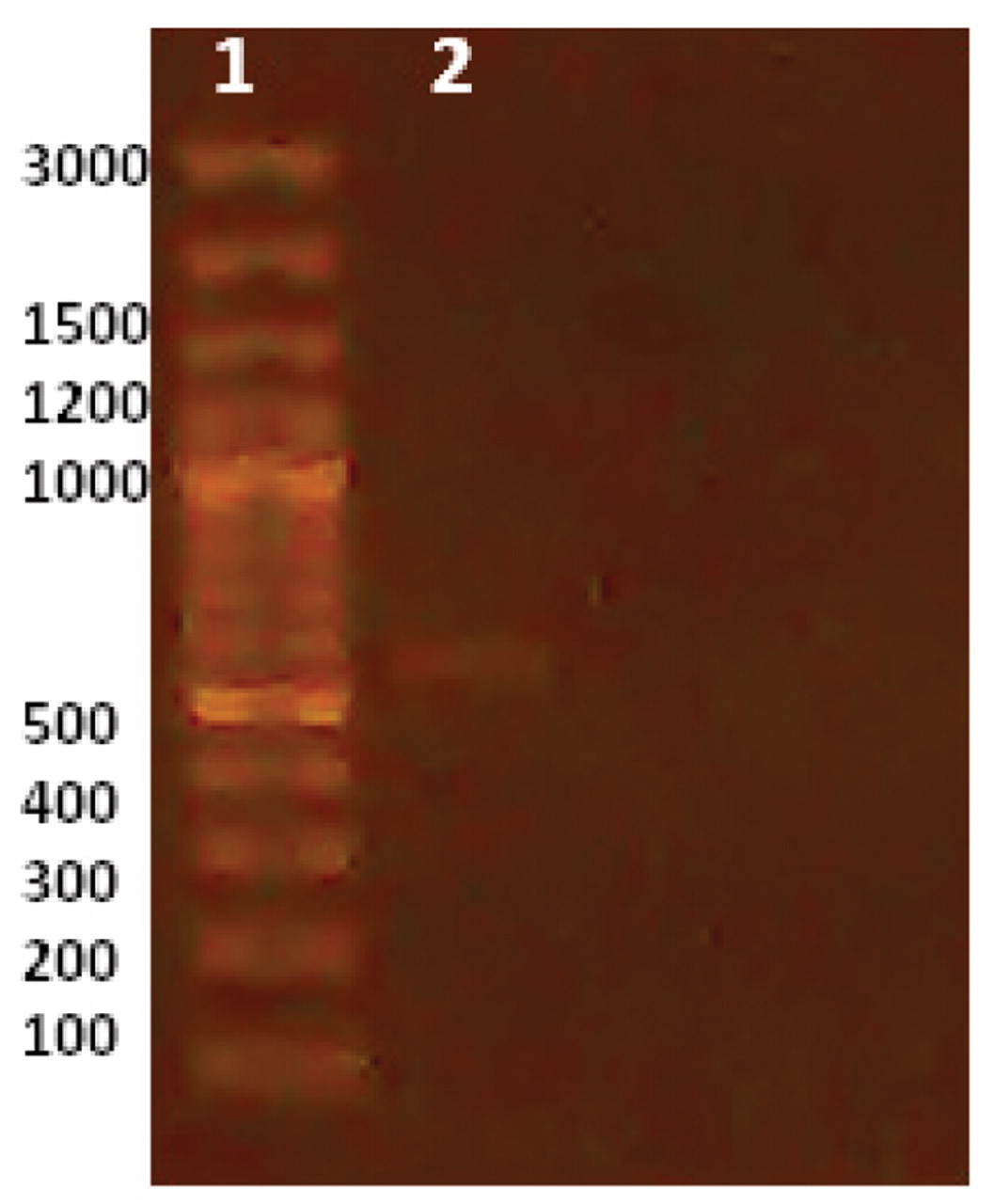
Other strains and species of pharmaceutical contaminants were used for examination of used primers specificity. The results are shown in Table II and indicate that the primers used are very specific for each microorganism and no interference or cross-reactivity were observed. To validate the influence of the enrichment step on improvement of PCR sensitivity for contaminants detection, bacterial inocula of each microorganism were prepared at 106 CFU per milliliter or gram and serially diluted to reach 100 (1 CFU) per milliliter or gram. PCR was carried out for these concentrations and after enrichment at 35 °C for 18 h. The minimum limit for detection of the four pharmaceutical contaminants in enriched media was 100 CFU/mL of original sample for all inoculated microorganisms (Figure 3), while in un-enriched media, a barely detectable PCR band for E. coli was detected at level of 106 CFU/mL (Figure 4) and no bands were detected for the other three microorganisms. In addition, PCR results were reproducible using another thermocycler (Techne TC-412, Staffordshire, UK) and reagents (Taq PCR master mix kit, QIAGEN, Hilden, Germany) confirming the robustness of the assay.
Specificity of the Primers and Lack of Both Interference and Cross-Reactivity

Effect of enrichment. A: Salmonella, B: E. coli, C: Pseudomonas, D: S. aureus. Lane 1: marker, Lanes 2–8: 106 pre-enriched inocula (Lane 2), serially diluted to reach 100 (Lane 8).

In un-enriched media, a barely detectable PCR band for E. coli was detected at level of 106 CFU/mL, and no bands detected for the other three microorganisms. Lane 1: marker, Lane 2: 106 inocula.
Sequence Analysis
The sequencing results showed 100% identity to sequences of the corresponding genes tested available at the gene bank for several species of each genus. This shows the high conservation of used primers sequences to their related microorganisms.
Discussion
The number of published studies demonstrating the application of rapid molecular methods such as PCR in the pharmaceutical industry reflects the importance of such techniques. Detection of microbial contamination in pharmaceuticals by conventional methods depends mainly on morphological and biochemical characteristics of tested microorganisms; they are time-consuming, as they require incubation times lasting between 3 to 5 days, and require large amounts of media. Furthermore, the recovery of microorganisms in tested pharmaceutical samples is affected by the presence of included preservatives and/or bacterial injury due to some pharmaceutical manufacturing processes. The main advantage of the PCR is the quicker turnaround time, allowing rapid corrective actions and, consequently, avoiding economic losses. Also, other advantages can be achieved if the raw materials are limited or need several steps of synthesis.
In this study the target bacteria were E. coli, P. aeruginosa, S. aureus and Salmonella spp, as specified bacteria according to pharmacopeial recommendations. Uniplexed PCR was performed for detection of each microorganism, individually targeting the conserved region in each bacterium genome. Further optimizations were done to perform duplex and multiplex PCR assays considering relative concentrations of competitive primers used in the reaction (40). The uniplexed PCR amplicons were successfully sequenced, confirming the conservation of used primers. Other validation parameters such as specificity, sensitivity, and robustness were well examined.
Sample pre-enrichment prior to DNA extraction is an important step because direct detection of contaminants by PCR assays cannot be applied to pharmaceutical samples for many purposes. Pre-enrichment has the advantage of increasing the reaction sensitivity by multiplication of target microorganisms to reach detectable limits, and this is due to the fact that the microbial requirements in the pharmaceutical industry necessitate the choice of raw materials of high quality and application of controlled production processes, so the microbial bioburden in case of microbial contamination is expected to be low. Enrichment provides dilution of PCR inhibitory substances in pharmaceuticals, especially products that consist of complex matrices. Another advantage of enrichment is the dilution of dead cells because PCR can not distinguish between viable and dead cells; however, this could be overcome by digesting the DNA and targeting the RNA using reverse transcriptase polymerase chain reaction (RT-PCR) (41) or the use of reagents such as ethedium monozide or propidium monoazide to be included in PCR assays for differentiation between these cells (42), but their suitability and application for use on a wide range of microorganisms is still under debate.
In this study, PCR was able to detect the contaminants in artificially contaminated samples of different dosage forms and chemical compositions, and with initial bioburden as low as 100 CFU per milliliter or gram. The total time for microbial detection was reduced to 27 h, compared to time consumed by using conventional methods. A successful recovery of inoculated microbial mixture was achieved by enrichment in non-selective media (TSB) as recommended by Casey et al. (43) and according to USP 34 〈62〉 (11). The suitable criteria of enrichment can be determined by enriching of positive control; if not successful, other modifications can be adopted to achieve recovery: further dilution, using neutralizers, membrane filtration, or combination of these methods.
In this study, when performing uniplex PCR, no difference in resulted bands was observed between DNA extraction by simple cell lysis as performed by Jimenez et al. (29) and Samadi et al. (36) or commercial spine column kit. The first extraction method is considered to be cost-effective by provision of in-house prepared reagents, but it does not provide sufficient purified DNA such as the second method, which was suitable for duplex and multiplex PCR. Also, successful multiplex PCR was performed by Karanam et al. (37) using phenol chloroform extraction, which is inexpensive. However, it is considered to be hazardous and inhibitory to PCR reactions in some instances (44). The use of lysis buffer containing TRIS-EDTA, Tween 20, and Protinase K, or the presence of lysozyme as a component of a commercial kit ensures an efficient extraction of Gram-positive S. aureus in artificially inoculated samples and does not affect the results concerning the other inoculated Gram-negative bacteria (29, 45).
Simplification of PCR assays can be achieved by employing user-friendly tablets such as the BAX™System (DuPont, Wilmington, DE, USA) (28) or ready-to-use beads such as the Ready-To-Go System (Pharmacia Biotech, Piscataway, NJ, USA) (29⇓–31), allowing minimized sample preparation and ease of avoiding cross-contamination. Furthermore, several features of thermocyclers used nowadays contribute to the use of PCR in routine work: as the gradient thermocycler, allowing the simultaneous screening of pharmaceutical samples, using primers with different annealing temperatures and resulting in detection of different microorganisms, in a single PCR run, without compromising efficacy. A previous work performed by Jimenez et al. (33) used RoboCycler 96 gradient (Strategene, La Jolla, CA, USA). Other companies such as Techne (Staffordshire, UK) and Biorad (Hercules, CA, USA) adopted manufacturing of this type. Recent developments achieved in thermocycler design include considering the number of wells and blocks leading to increased capability of analysis of a high number of samples independently.
Bioinformatics provides efficient tools for determining appropriate primer sequences, structural analysis (hairpin, self, and heterodimer formation), specificity assessment, suitable Tm, and sequence analysis. Advanced PCR formats can be applied for detection of contaminants quantitatively by real-time PCR (32, 34). The use of PCR in microbiological assessments of medical devices has been also reported (39).
The use of the four bacteria as indicator pathogens does not neglect that other bacteria might be problematic in microbiological evaluation. Depending on the DNA sequence homology between species and with the aid of bioinformatics tools and published studies, we may widen the investigation field to include other microbes whether pathogenic or spoilage species considered objectionable such as Burkholderia (30), consequently saving time and allowing a high-throughput screening of a wide range of targets (46).
The use of PCR technology in the pharmaceutical industry field to substitute for conventional microbiological methods is governed by considerations of new technology transfer, which include setting up of PCR laboratory considering the separation of different PCR activities, whether by place or time, and the use of appropriate equipment and reagents. Preparation of well-trained personnel is a critical point and can be achieved by provision of a team of experts. Furthermore, construction of quality control/quality assurance systems to provide standard operating procedures and worksheets to control each step performed in this technique is essential (47).
The application of new methods faced some obstacles due to their high cost, hesitation of managers to try new methods, lack of understanding of these technologies, and lack of abundant resources of validation and technical support. Fortunately, the Parenteral Drug Association (PDA) provided a technical report that outlined the evaluation, validation, and implementation of new alternative microbiological methods (48). In addition, USP 34 (11) has provided comprehensive discussions of rapid molecular methods, including terminology of different related aspects 〈1125〉, extraction, detection and sequencing 〈1126〉, amplification 〈1127〉, microarray 〈1128〉, genotyping 〈1129〉, detection of trace nucleic acids 〈1130〉, and validation of alternative microbiological tests methods 〈1223〉. This may encourage microbiologist to use a nucleic acid–based techniques as alternative methods in this field. Although PCR is applied for the detection of mycoplasma in cell cultures, its use as an efficient tool in detection of indicator pathogens in pharmaceuticals is still limited to academic and research settings, as most of published literatures concerning this topic were carried out in research centers, academic institutes, or research and development department of some companies such as GlaxoSmithKline (formerly Block Drug Company), where Jimenez et al. performed their previous work (22⇓⇓⇓⇓⇓⇓⇓⇓–31, 33).
Conclusion
The detection of bacterial contaminants in pharmaceutical preparations has been revolutionized recently by the availability of techniques such as the use of PCR, which is gaining huge momentum. In the Nile Company for Pharmaceuticals and Chemical Industries, where the authors performed this work, the PCR technique for the detection of four specified bacterial contaminants that are usually checked for in oral or topical product have been optimized. Optimization of this technique, in one of the main pharmaceutical companies in Egypt, can serve as a beginning of applying these molecular tests in quality control departments in Egypt and the Middle East, with the consequent benefit of saving time accompanied by rapid release of pharmaceutical products in such a demanding market.
Conflict of Interest Declaration
The authors declare that they have no competing interests.
Authors' Contributions
SMR carried out the experimental procedure and participated in drafting the manuscript. ASY supervised the experiments, analyzed the resulting data, and wrote the manuscript in its final form. MAA conceived and supervised the study. All authors read and approved the final manuscript.
Acknowledgments
The authors would like to thank Dr. Yasser Abdelrahman at the Microbiology and Immunology Department, Faculty of Pharmacy, Cairo University, for his efforts in the early stages of this work.
- © PDA, Inc. 2012




{kind=link}
{kind=link}
{kind=link}
{kind=link}