
Abstract
The assurance of sterility of a parenteral drug product, prior to any human use, is a regulatory requirement. Hence, all strategies related to container closure integrity (CCI) must demonstrate absence of microbial contamination through leaks as part of the container closure system (CCS) qualification, during manufacturing, for quality control purposes and to ensure microbiological integrity (sterility) during storage and shipment up to the end of product shelf life. Current regulatory guidances, which differ between countries and regions, provide limited detail on how to assess CCI. The new revision of USP <1207> aims to provide extensive and detailed guidance for CCI assessments for sterile products. However, practical questions and considerations are yet to be addressed by the pharmaceutical industry. These may include: (1) choice of method, for example whether a deterministic CCI method (e.g., helium leak) is preferable over the probabilistic CCI method (e.g., microbial ingress), (2) the type of primary packaging (e.g., vial, syringe, device), (3) dosage form (e.g., liquid versus lyophilisate), (4) suitable acceptance criteria, (5) appropriate sample size, (6) the most appropriate way to introduce artificial leaks into the CCS, (7) ensuring suitable assurance of CCI during drug product manufacturing, and (8) evaluating CCI under intended shipment and storage conditions (e.g., in the frozen state).
A group of European industry peers have met to discuss these and other related questions in order to provide their viewpoint and best practice on practical approaches to CCI. Their perspective is provided in this white paper. Through these discussions, it became clear that there is currently no gold standard for CCI test methods or for the generation of artificial leaks; therefore flexibility toward CCI approaches is required. Although there should be flexibility, any CCI approach must consider the intended use (e.g., CCS qualification, routine manufacturing, or quality control) and product design (e.g., primary packaging, liquid versus dried product).
LAY ABSTRACT: The assurance of sterility of a parenteral drug product prior to any human use is a regulatory requirement. Hence, all strategies related to container closure integrity (CCI) must demonstrate absence of microbial contamination through leaks as part of the container closure system (CCS) qualification, during manufacturing, for quality control purposes and to ensure microbiological integrity (sterility) during storage and shipment up to the end of shelf life. Current regulatory guidances, which differ between countries and regions, provide limited detail on how to assess CCI. The new revision of USP <1207> aims to provide extensive and detailed guidance for CCI assessments for sterile products. However, practical questions and considerations are yet to be addressed by the pharmaceutical industry.
A group of European industry peers have met to discuss these and other related questions in order to provide their viewpoint and best practice on practical approaches to CCI. Their perspective is provided in this white paper. Through these discussions, it became clear that there is currently no gold standard for CCI test methods or for the generation of artificial leaks; therefore flexibility toward CCI approaches is required. Although there should be flexibility, any CCI approach must consider the intended use (e.g., CCS qualification, routine manufacturing, or quality control) and product design (e.g., primary packaging, liquid versus dried product).
- Container closure integrity testing
- Methods for container closure integrity testing
- Artificial leaks
- Industry perspective
- Container closure system
- Primary packaging
- USP <1207>
Introduction
In response to the revision of USP <1207>, Packaging Integrity Evaluation—Sterile Products (1), members of several companies within the pharmaceutical industry have come together to address their common understanding of the application of container closure integrity testing (CCIT). The purpose of this paper is to further raise awareness, throughout the industry, of the complexity of topics when evaluating leaks in container closure systems (CCS). The information contained herein reflects the experiences of the contributing companies and is not regarded to be exhaustive of the industry. Container closure integrity (CCI) addresses the maintenance of integrity to prevent microbiological ingress in sterile product packaging until the time of use (e.g., product opening for single-dose products). All products labeled as sterile are expected to be free of microbial contamination throughout their shelf life. Out of the scope of this paper are other considerations for CCI that may include tightness against exchange of gas, water vapor, or volatile substances through leakage or permeation into or out of the CCS. The main points of consideration related to CCIT are summarized below:
There is currently no universally accepted test method or gold standard for conducting CCIT. Worldwide and local regulatory requirements offer no clear distinction as to what is required other than that sterility shall be maintained until the end of product shelf life. Local regulations may differ and should be considered as well as experience within an organization.
Artificial leaks do not necessarily simulate actual defects due to, for example, irregular shapes and pathways in a CCS. There is also high variability depending on the method used to create and measure such holes. Artificial leaks also cannot be easily related to a leak size.
Different CCIT methods can serve the same purpose when appropriately qualified.
There is a need to correlate microbial contamination and the physical CCI (pCCI) test method.
CCIT is conducted to assure the integrity of a CCS over the life cycle of a product. This includes initial product development, clinical manufacturing, establishment of shelf life, and commercialization of a product. The revised USP <1207> describes historical reasons for conducting CCIT, which references a tragic incident where approximately 10% of patients died due to a package integrity failure. This led to contaminated intravenous (IV) fluids and caused an estimated 2000 to 8000 cases of infection in the bloodstream (2). This example highlights the responsibility of the pharmaceutical manufacturer to ensure product quality and safety, and CCIT is an important aspect required to verify this. Various European directives, monographs in pharmacopoeias, U.S. Food and Drug Administration (FDA) Guidance to Industry, and International Council on Harmonization (ICH) guidelines have been published to mandate the expectation toward industry in which CCS qualification and the validation of critical phases during the manufacturing processes must be achieved to meet final product specifications throughout the product's intended use (1, 3⇓⇓⇓⇓⇓⇓⇓⇓⇓–13). However, these regulations and guidance do not provide specific details regarding how CCIT shall be conducted.
Methods for CCIT
There are several methods available for pCCI testing. They can be classified into deterministic and probabilistic methods. Definitions for this terminology, below, are taken from the revised USP <1207>:
Deterministic method: A method in which the leakage event being detected, or measured, is based on phenomena that follow a predictable chain of events. In addition, the measurement of leak detection is based on physicochemical technologies that are readily controlled and monitored, yielding objective quantitative data (1).
Probabilistic method: A method that is the converse of a deterministic leak test method, being stochastic in nature. Probabilistic tests rely on a series of sequential and/or simultaneous events, each associated with random outcomes described by probability distributions. Thus, the findings are associated with uncertainties that necessitate sufficiently large sample sizes and rigorous test-condition controls to obtain meaningful results (1).
The most commonly used deterministic and probabilistic methods for CCIT are shown in Table I. This table describes CCIT methods, their purpose, the advantages and disadvantages commonly stated in literature for particular types of closure system, and actual industry experiences from the authors. Both probabilistic and deterministic methods have been applied successfully for CCIT throughout industry and, if adequately qualified, are justified and widely accepted. The information in Table I demonstrates that there is currently no single method that can be considered the “best” way to assess CCS integrity of a drug product within a specific stage of the drug product life cycle. In addition to the lack of a gold standard, there is currently no universally valid way of creating artificial leaks (see section 1. Introduction of Artificial Leaks) or a single value or default range for acceptance criteria.
Examples of pCCI Techniques for Consideration for CCS Qualification, Manufacturing Process Control, and/or Quality Control
Any test method should be suitable for its intended use (see General Considerations for the Applicability of CCIT) and may significantly differ for a given CCS at different stages of the product life cycle. For example, during development of a primary package, a low throughput but highly sensitive test method (e.g., helium leak detection) may be the method of choice in order to support the CCS selection. In support of the manufacturing process monitoring activities for a specific drug product, a test method with higher throughput but lower sensitivity, such as vacuum decay, dye ingress, or high-voltage leak detection (HVLD), may be more appropriate in order to detect defects created during manufacturing of a product.
Maintaining the integrity of a CCS can be considered an obligatory critical quality attribute (CQA) for a quality by design (QbD) approach, in which it is evaluated throughout development and manufacturing. In practice, a series of different pCCI methods could be used throughout the product life cycle based on the level of understanding of the primary packaging components in combination with the drug product formulation and critical drug product manufacturing process unit operations and parameters. The purpose of the CCIT method is to have a strong level of understanding of the primary packaging components, process parameters, and control over operations. Moreover, the acceptance criteria for a given CCS can vary (be tightened or widened) over its life cycle based on the level of knowledge gathered throughout development and process validation and continuous process verification.
Each pCCI method has a different sensitivity (associated with specific test conditions) and different acceptance criteria (pass/fail threshold) that need to be established and verified for a specific CCS. It is appreciated that a risk-based approach can justify leveraging of generic settings for a specific CCS to also include changes in product content and thereby obviate the need for frequent revalidation. Once a representative CCS is established and validated, the CCIT results and/or methods can be used for different product contents. However, a robust change control of any validated process must be maintained. It should be noted that product sterility is a requirement for any stability program for sterile products, at least at release and at the end of shelf life. Therefore, sterility testing and CCIT data are both provided according to various best practices depending on the target regulatory market (9).
The following criteria are considered important for the selection of any pCCI method:
the intended purpose, for example CCS development and qualification, manufacturing process control or validation, or stability testing
prior knowledge for the development of the CCS, for example initial product development with a CCS versus further development of a CCS for a new product
the CCS format, for example vial, syringe, drug/device combination product, IV bag
the CCS material, for example flexible, glass, polymer
the type of product, for example liquid versus lyophilisate, small versus large molecule, water-based versus solvent or oily formulation, conductivity, viscosity, gas headspace composition, ambient pressure or vacuum/overpressure
test duration
the required sensitivity
the type and availability of samples with artificial leaks
the sample size required for a specific study
need for sample preparation and potential risks associated with the sample preparation (e.g., label removal, vials to be emptied and cleaned)
In many cases, the initial selection of the test method is based on the previous experiences with a specific pCCI technology. Knowledge for applying various methods over the drug product life cycle come from company experience, experience with similar products or packages, published literature, or standards and guidance documents (see Table I).
Additionally, the following key attributes should be considered for method selection:
destruction or non-destruction of the test sample
inline, online, offline, or nearline
sampling or 100% testing
testing efficiency, which includes testing speed (i.e., measurement time per unit) and throughput (includes rest times for sample equilibration and measurement of control groups)
testing frequency and schedule: only tested once or multiple times, for example as part of a stability program (considering the potential impact of gas permeation)
costs associated with the purchase, validation, and maintenance of equipment as well as maintaining the environmental conditions
suitability for testing a filled or empty CCS
independence from physicochemical characteristics of the CCS content
performance characteristics: sensitivity (i.e., limit of detection), method robustness, precision, and accuracy
output of test result: pass/fail (i.e., presence or absence of a leak above a certain threshold), location of the leakage, or the size or flow rate of a leak
risk of equipment contamination when exposed to a leaky sample.
In all cases, the equipment used must be appropriately calibrated and qualified and the test methods suitable for their intended use. Test methods should include suitable positive controls (with known leakage) and negative controls (with no intentional leakage) for the system suitability test (SST). For “probabilistic” methods a larger number of control samples is often required due to the nature of the method. It may be considered permissible for some of the multiple positive control samples to exhibit no leakage during SST due to its probabilistic nature. The test conditions of “probabilistic” test methods such as dye ingress have parallels to microbial ingress, as both can be executed under similar vacuum/pressure challenge conditions and both are based on probabilistic events of contamination/ingress. In contrast to this, the quantitative test results (e.g., helium leakage, oxygen content, or pressure decay) need to be correlated to a pass/fail threshold through method development and validation, which can be challenging to define.
In addition, there are several types of CCIT methods that do not directly measure leakage but are complementary because they indicate seal quality and support CCS characterization. Examples are burst testing, residual seal force (RSF) testing, pull-off force testing, X-ray computed tomography (CT), and torque testing. Seal quality methods support individual aspects for evaluating the system throughout any part of the product life cycle.
Introduction of Artificial Leaks
Samples with artificial leaks (holes or channels) are used as positive controls to assess the capability of a leak test method. They should be created for any CCIT method to ensure appropriate method development, evaluation, and verification prior to routine testing. There are various ways of creating leaks, and the most commonly used methods are listed below:
laser drilling into the body of the container
laser drilling into a metal plate or tubing that is integrated to a CCS
micron wires inserted at the interface between the closure and container
micropipettes (glass) inserted into the stopper or glued into an artificial hole in the container
capillaries (fused silica, nickel, glass) inserted into the stopper or glued into an artificial hole in the container.
Each of these preparations has its own challenges, and in order to define a leak, the appropriate acceptance criteria are required. Some of the challenges associated with creating an artificial leak, using the methods listed above, are presented in Table II.
Advantages and Disadvantages of Different Methods for the Introduction of Artificial Leaks
There is currently no specific standard to prepare a leak and determine its size once integrated into the CCS and, moreover, there is no need to achieve an absolute reference to a leak size, for example 0.2 μm, or a universally acceptable minimum flow rate. The point is that there should be enough flexibility when establishing and qualifying a CCIT method so that each company can justify its test method. If an absolute reference is not used, then a relative reference, such as a correlation to microbial ingress for a given artificial leak size, may be used (14). This flexibility includes the method used, setup details, and conditions of testing that include the type of artificial leak samples and associated procedures to create them.
The detected leakage rate is often converted and expressed in terms of hole size for easy conceptualization of the degree of defect, yet artificially created leaks, such as laser-drilled holes or micron wires, are dimensionally irregular. Currently there is no pCCI method available that measures a hole size, although some do measure leakage rate (e.g., helium leak testing utilizes a volumetric flow of gas). It is important to consider that a leakage flow rate does not necessarily correlate to a specific hole size, as discussed below. Moreover, imaging of artificial leaks by microscopy can be challenging at best (15).
There will likely be variability in positive controls due to the method of forming the leak, and repeatability, or the precision, between samples should be part of method validation. Ideally a hole or channel would be circular, having a certain diameter and length. However, the created artificial hole is likely to be irregular in shape, for example having variations in length, inner diameter, and circularity of its openings. This could potentially change over time or during use. For example, a micrometer-sized hole could become blocked with particles, a hole in glass may increase over time, or a tortuous/asymmetric path may be present. The reality is that micrometer-sized defects often present themselves as tortuous paths of varying diameter, typically found between closure and container. For example, vial defects may include glass cracks or breaks, misaligned closures, and poorly crimped seals (16, 17). There is also an assumption that there is only one hole present in a sample whereas in reality a series of holes or pathways may exist. Referring to leak size in micrometers may suggest an absolute unit that can be compared to the size of bacteria, but this assumption is misleading. It has been reported that microbial ingress starts to occur anywhere between 0.3 and 20 μm, so no consistent critical leak size, or absolute threshold, exists (14, 20, 21, 26). Leak size alone will not determine if bacteria may ingress into a CCS. Other factors apply such as leakage pattern, for example path length and geometry, and environmental conditions, presence of liquid in the leak path, as well as test conditions, for example microbiological test organisms used, their concentration, incubation time, and test parameters.
Leakage dynamics are different for different defect types and packaging materials. The inclusion of defects positioned above and below the liquid fill level is important if the leak test method's performance is a function of liquid or gas presence in the leak path. Positive control sample populations should include small and large defects to represent the full range of anticipated leak sizes (16, 17) during test method development and qualification. An artificial leak representing the acceptance criteria should be used at minimum during routine testing.
There are various types of techniques available for creating leaks and each can pose sample-to-sample variation. A fused silica capillary could become partially occluded during testing or preparation due to the multiple manipulations such as bending, cutting, and gluing that would result in a partial or complete blockage. Also, silicone oil in prefilled syringes may block artificial leaks (e.g., laser-drilled holes). Alternatively, a void may become wider during use or from storage and transportation, which would result in false sensitivity. Examples include glass micropipettes that are very fragile, with the potential for the tip to break, or laser-drilled holes (in a glass container), which may widen due to tensile fields within the glass.
It should be noted that a nominal leak size may actually be different in terms of volumetric flow and hence represent a different cut-off size (the point of failure, e.g., for bacterial contamination). A capillary with a non-negligible length (e.g., fused silica capillary) may be equivalent to an orifice of negligible length with a much smaller nominal diameter. For example, a 10 μm fused silica capillary of 12 mm length would exhibit a laminar air flow of approximately 0.0034 mL/min (dry air at a differential pressure of 1013 mbar versus an outlet pressure of 1.3 mbar at 20 °C, 5.77 × 10–5 mbar*L/s). This is calculated in this way:
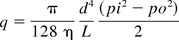 η = viscosity of the air at 20 °C (18.19 × 10−6 kg/m*s), L = length of capillary in cm, d = internal diameter of capillary in cm, pi = inlet pressure in mbar and po = outlet pressure in mbar, and q = flow of air in mbar * L/s.
η = viscosity of the air at 20 °C (18.19 × 10−6 kg/m*s), L = length of capillary in cm, d = internal diameter of capillary in cm, pi = inlet pressure in mbar and po = outlet pressure in mbar, and q = flow of air in mbar * L/s.
An orifice of approximately 0.63 μm with negligible length would have the same volumetric flow rate (assuming laminar flow). This is according to Lenox Laser orifice sizing calculator using same pressure settings for air at 20 °C and a flow rate of 0.0034 sccm.
It is therefore recommended that each pCCI method and its measuring unit should be correlated with a specific relative (leak or flow rate) reference instead of relying solely on the hole size (15).
Considerations of Microbial Challenge Container Closure Integrity Testing (mCCIT) To Establish Acceptance Criteria for Physical Container Closure Integrity Testing (pCCIT)
A sterility test is a pharmacopeial test method used to determine that the CCS has maintained product sterility at any single point in time. Hence, it is typically used at the start and end of stability testing for a product's shelf life. In contrast to this method, mCCIT is a leak challenge test performed on containers filled with nutrient medium. A typical mCCIT method involves submersion into a predefined microbial suspension for a fixed period of time under controlled conditions. This condition could be considered the worst-case scenario because the primary package is surrounded by a high concentration of small living microbes, and ingress would occur naturally if given enough time and a liquid-filled leakage path is present. Apart from the need to define an adequate immersion period, the use of underpressure and/or overpressure can add an additional driving force for microbial ingress as well as simulate environmental changes that may be encountered during transportation. Note that there are currently no standard time and pressure parameters established for mCCIT, however, ASTM D4169 (22) and D6653 (23) can be referenced when simulating transportation conditions. Furthermore, microbe selection, microbial concentration, or growth media also lack standards, but guidance may be found in published literature (24).
A mCCIT is not a measure of leak rate, but it is one way of supporting the establishment of acceptance criteria for pCCIT based on experimental data during initial evaluation of a CCS (15, 20, 21, 25, 26). To correlate both methods, an mCCIT method can be used with similar artificial leaks as the pCCIT method for a specific leak rate or hole size. Then, the acceptance criteria for the pCCIT method can be based on a mCCIT study. Other ways to establish the acceptance criteria for a pCCIT is to reference established literature (14, 15, 20, 21, 26) or company-based studies with comparable CCSs if a suitable justification is provided. According to the experience within this group of authors, a universal way for setting acceptance criteria based on the probability of microbial ingress does not exist, for example whether 1% or 100% of the samples show ingress in the study for setting the acceptance criteria. Hence, justification is required for the rationale to set acceptance criteria based on a predictable leak rate and microbial contamination. Advantages of using a pCCIT method are the greater consistency, ease of use, speed of testing, and lower cost in leak detection compared to mCCIT.
General Considerations for the Applicability of CCIT
The application of CCIT warrants different considerations based on the dependence on the CCS or product, and different methods may be appropriate depending on the stage of the product life cycle. The manufacturer needs to have a full understanding of the test method capabilities for each of its CCIT techniques.
Highly sensitive methods may be the best choice during early development to qualify a CCS and characterize critical process parameters during product manufacture, where a lower throughput is acceptable. In production, a less-sensitive method is justifiable because of the need to detect process- or component-related issues. However, the sensitivity of a chosen pCCI method should be justified and match the intended purpose. Alternatively, a risk-based assessment could be performed taking into account the appropriate level of CCIT detection capabilities required for a particular CCS type.
Careful consideration should be given to the type of CCIT procedure used for specific CCSs, such as in the case of combination products and devices. For example, if a vacuum decay test is established for a syringe-based CCS, the detection limit may increase for the same CCS assembled with a needle safety device or in an autoinjector. A lower sensitivity may be warranted to have a test method with repeatable results in order to assess the impact of assembly risks to the primary packaging integrity.
The sample size used for testing should be sufficient to provide adequate assurance of CCI and varies on the basis of (i) the complexity of the product-package configuration, (ii) the specifics of the user specification requirements, and (iii) the prior experience within the company for similar CCS (1).
CCIT in the Product Life Cycle
There are several areas of application for CCI testing, which include the following:
Primary packaging development and CCS qualification
Drug product manufacturing process characterization and validation
Drug product routine manufacturing over the entire product life cycle (clinical to commercial)
Evaluation of transportation and storage considerations
Drug product quality control testing, for example stability
The initial CCIT methods and acceptance criteria applied during packaging development (i, ii) may not necessarily be the same as those used in the other phases (iii–v) of the CCS life cycle. A risk assessment, including worst-case assessment of component variability and critical process parameters affecting the closuring process, can be leveraged in a QbD approach because CCIT can be used as part of the control strategy (16). For example, RSF and CCI testing can be applied during equipment qualification and manufacturing process characterization to evaluate minimum and maximum capping pressure and other crimping parameters (25, 27).
CCS qualification may be required only once per CCS combination and can be performed on an empty CCS or filled with a surrogate (e.g., with water or placebo). The CCS qualification would be applicable to various products in the same configuration. Transportation studies would not be performed with an empty but with a filled CCS in order to mimic realistic conditions related to the weight of the product and the vapor pressure of the contained solution. Even in this case, a surrogate may be appropriate.
Further considerations during development could include an assessment of component dimensional variability and its impact on CCI. Such an understanding could be leveraged for the same CCS for different products. A tolerance analysis of the dimensions of a container-stopper combination could be considered. However, actual testing of extremes is rarely possible because appropriate materials may not be obtainable. Hence, this type of analysis should follow a risk-based approach and is considered on a case-by-case basis.
CCS qualification does not need to be repeated for every product. However, CCIT can be part of the stability program for any drug product. Each manufacturer needs to justify and define CCIT methods and time points for stability testing, whether on validation or registration/commitment batches. Time zero and end-of-shelf-life testing may be extended with annual interim time points (9). Although pCCI can replace sterility testing in stability studies at time points other than release for the US (as per FDA guidance), sterility testing maintains a mandatory requirement for many non-US markets and countries at least at the end of shelf life. Yet, it should be well-understood that an acceptable CCIT assessment cannot be achieved by sterility testing alone because it does not challenge the CCS.
Further considerations include the assessment of the impact that transportation can have on the product, whether it is from breakage or stopper movement that could cause a sterility breach. CCIT is required as part of the transport validation in regulatory submissions for several countries such as Australia, Canada, and the US. Specific considerations should be addressed for products that require refrigerated or frozen storage and shipping conditions. In such cases, special attention is required to appropriately select the type of materials for the product packaging, including consideration of glass transition temperatures for plastic and rubber components (15). There should be documented assurance that the CCS integrity is not compromised by transportation or in the case of intended storage at extreme temperatures (e.g., below 0 °C or above 25 °C). CCI could also be assessed at excursions outside of intended storage temperatures. Industry continues to study these situations and evaluate foreseeable conditions using new applications of technology to assess any compromise to CCI and seal quality (17, 22, 23, 28⇓⇓⇓–32).
In the case where a 100% CCIT strategy is implemented in manufacturing operations, a non-destructive method is required. In such instances the acceptance criteria may need to be appropriately adapted to achieve a realistic throughput and to minimize rejection of false positives (i.e., mistakenly rejected as leaking). When the CCS is suitably qualified and the closuring process is validated, defects that occur during well-controlled routine manufacturing process are expected not to occur or to be larger than the microdefects accounted for during qualification. This justifies reducing sensitivity at the line. Yet, this justified sensitivity for 100% CCIT inspection raises doubt of its application because it will not improve product quality or safety sufficiently to justify its costs and maintenance. Instead of performing a 100% CCIT of each batch, a well-controlled and validated manufacturing process (holistic approach) (28) may even be a preferred approach. Other controls, which are generally performed for parenteral products, such as 100% visual inspection and acceptable quality level (AQL) sampling, aim to further aid in the detection of critical CCI defects ranging from cracks to crimping, stoppering, or other assembly-related defects (16). However, some defects may not easily be detectable during visual inspection, for example those under the crimp cap, yet still be difficult to detect in a 100% online process. Therefore, each manufacturer should be left to define and justify their CCIT program and to what extent and frequency it is based.
Conclusion
In light of the revised USP <1207>, it is anticipated that there will be more scrutiny regarding the topic of CCIT and new regulatory expectations that need to be met by companies involved with sterile drug product manufacturing. This paper provides a reflection from within the industry on various practical aspects and approaches. Essential to industry is that there remains a degree of flexibility regarding the selection of CCIT methods and definition of acceptance criteria. There are no gold standard methods that are always superior to others nor is there a preferred, single way for creating artificial leaks. Leak holes described in terms of micrometers is an oversimplification and additional parameters need to be considered. Given the limitations of each technique to create artificial holes, a rationale on how to derive pass/fail thresholds is required. The various CCIT instruments currently available on the market each have their advantages and disadvantages. Hence, it is the responsibility of any pharmaceutical manufacturer to select and justify the selected test methods and to define appropriate acceptance criteria based on the intended use.
It is important to apply methods that are suitable for any given product/CCS combination and to ensure integrity through the product shelf life. A risk-based approach may assist in identifying an appropriate CCIT. Any CCIT must include the use of positive controls and consider the aspect of potential microbial contamination. A QbD approach to utilize CCIT can be applied to product development, process validation, and manufacturing controls. The strategies need to ensure product sterility for the products in its primary packaging and as combination products.
Manufacturers need to consider the relevancy of global market regulatory requirements. However, expectations regarding the CCIT strategy currently differ between countries and market regions due to different expectations from health authority agencies. Because a universally agreed CCIT guidance or international standard does not exist, there remains an essential need for manufacturers to apply flexibility and sound justification regarding their CCIT program, which is an obligation prior to the first human use of any sterile product.
Conflict of Interest Declaration
The authors declare that they have no competing interests.
Acknowledgments
The authors would like to acknowledge the following people who contributed to the development or review of this work: Rene Spycher, Janssen R&D; Valeria Delia, Merck Serono; Ingeborg Kraemer Pittrof, Roche; Nathalie Yanze, Genentech; Joerg Zuercher, Bayer; Pierre Guiswe, Boehringer Ingelheim; and Jacques Maring, CSLBehring.
- © PDA, Inc. 2017
References




Jump to section
- Article
- Abstract
- Introduction
- Methods for CCIT
- Introduction of Artificial Leaks
- Considerations of Microbial Challenge Container Closure Integrity Testing (mCCIT) To Establish Acceptance Criteria for Physical Container Closure Integrity Testing (pCCIT)
- General Considerations for the Applicability of CCIT
- CCIT in the Product Life Cycle
- Conclusion
- Conflict of Interest Declaration
- Acknowledgments
- References
- Figures & Data
- References
- Info & Metrics
Related Articles
Cited By...
- Mechanical Container Closure Integrity Test: A Method for Cartridge Systems
- Comparing Container Closure Integrity Test Methods--Performance of Headspace Carbon Dioxide Analysis versus Helium Leakage Using Positive Controls
- Container closure integrity testing and process validation of closed system transfer devices for aseptic reconstitution of drug vials connected to fluid bags
- A Multicompany Survey Study for Helium Leak Container Closure Integrity Test
- Container Closure Integrity Test Using Frequency Modulation Spectroscopy Headspace Analysis with Carbon Dioxide as a Tracer Gas
- Determining Maximum Allowable Rubber Stopper Displacement for Container Closure Integrity (CCI)
- Quantifying the Vial-Capping Process: Reexamination Using Micro-Computed Tomography
- Single-Use System Integrity I: Using a Microbial Ingress Test Method to Determine the Maximum Allowable Leakage Limit (MALL)
- Comparing Physical Container Closure Integrity Test Methods and Artificial Leak Methodologies
- Container Closure Integrity Testing--Method Development for Freeze-Dried Products Using Laser-Based Headspace Oxygen Analysis
- Evaluation of Container Closure System Integrity for Storage of Frozen Drug Products: Impact of Capping Force and Transportation
- Sealing Behaviour of Container Closure Systems under Frozen Storage Conditions: Nonlinear Finite Element Simulation of Serum Rubber Stoppers