
Abstract
Drug products and medical devices can contain leachable impurities that could adversely affect patient health during their clinical use. To establish patient exposure to leachables, drug products and packaging, manufacturing system, or medical device extracts are analytically screened for leachables or extractables. For organic extractables/leachables, the screening process typically involves a chromatographic separation coupled with an information-rich detection method. Information contained in the detector response (e.g., the chromatographic peak) is processed to establish quantities and to elucidate identities for the detected compounds. Organic extractables and leachables screening methods and procedures have proliferated with little, if any, attempt at standardization, creating the situation in which virtually every testing laboratory has their own analytical testing and data processing methodology. This raises the possibility that two different labs screening the same extract or drug product would report extractables or leachables profiles that differ in the number of compounds reported, the identities of the reported compounds, and the extracted (or leached) amounts of the identified compounds. Although standardization of the screening methods and procedures themselves would reduce lab-to-lab variation, such an approach would be difficult to implement. Thus, standardization of the screening outputs by setting quality standards for the outputs is considered. For example, the method's ability to detect a broad cross-section of potential extractables/leachables is established by testing a test mixture of representative compounds. Additionally, this author proposes that reported compound identities should be confident to be used in safety risk assessment; use of lower quality identities requires that the lower quality be accounted for in the assessment, perhaps by use of an uncertainty factor. Similarly, it is proposed that reported concentrations should be semi-quantitative to be used in safety risk assessment; use of lower quality concentrations requires that the lower quality be accounted for in the safety risk assessment, perhaps by use of an uncertainty factor.
Introduction
Pharmaceutical drug products and medical devices minimize, mitigate, alleviate, or eliminate undesirable medical conditions in the human population. It is highly desirable that drug products and medical devices accomplish these objectives with minimal adverse effects. However, drug products can contain impurities, termed leachables, derived from the drug product's packaging system and/or manufacturing equipment. A patient who is administered the drug product will be exposed to such leachables, and through this exposure to the leachables might experience an adverse health effect. Alternatively, substances present in or on a medical device can leach from that device into the device's medium of contact with the human body during the device's clinical use. Exposure to such device-related leachables could also produce an adverse health effect.
To establish patient exposure to leachables, drug products, packaging, or manufacturing system extracts and medical device contact media or extracts are screened (i.e., nontargeted analysis) for leachables or extractables to establish whether leachables achieve levels sufficiently high that they must be identified and quantified.
Considering organic extractables/leachables, the screening process typically involves a chromatographic separation coupled with an information-rich detection method. Information contained in the detector response (e.g., the chromatographic peak) is processed to establish the detected compound's quantities and to elucidate identities.
Organic extractables and leachables screening methods and procedures have proliferated with little, if any, attempt at standardization, creating the situation in which virtually every testing laboratory has their own analytical testing and data processing methodology. Some degree of standardization happens by accident as experts naturally gravitate toward the best available science; for example, it is general practice that screening for organic extractables involves multiple chromatographic methods of orthogonal but complementary nature, such as gas chromatography (GC) and liquid chromatography (LC). Furthermore, mass spectrometry (MS), given its sensitivity, specificity, and identifying information content, is universally the detector of first choice. Thus, a preponderance of testing laboratories have both a GC/MS method and one or more LC/MS methods. Furthermore, some degree of unintentional standardization occurs due to the natural emergence of common practices in the analytical instrumentation or consumables or data processing algorithms. Thus, for example, electron ionization (EI) MS detectors for GC are standardized at an electron energy of 70 eV, and GC separations are generally accomplished using a common stationary phase (USP designation G27, 5% phenyl, 95% dimethylpolysiloxane) and either an internal database of mass spectra and/or the Wiley/NIST library of mass spectra is generally used for identification of GC/MS peaks via mass spectral matching. Given the greater number of operational parameters associated with LC analyses, a lesser degree of organic LC standardization has occurred, although most LC screening method are based on reversed-phase separations performed on C18 columns with binary organic/water mobile phase gradients.
Unintentional standardization notwithstanding, analytical screening methodologies for organic extractables and leachables vary widely from lab to lab in terms of their critical operations, critical operating parameters, and settings for the critical operating parameters. This lack of standardization in the analytical process has an unfortunate but predictable negative impact on the lab-to-lab agreement in the analytical results. That is, two different labs screening the same extract or drug product would possibly report extractables or leachables profiles that differ in the number of compounds reported, the identities of the reported compounds, and the extracted (or leached) amounts of the identified compounds. Such a possibility has been conceded in the E&L community of practice. For example, Zdravkovic (1) recently noted that “it is an unfortunate reality that an E/L screening study at multiple labs will, more often than not, yield datasets that are unaligned to at least some degree”. I myself have suggested that “if two practitioners use generally similar methods with different operating conditions to generate a chromatogram (which is the basis of an extractables or leachables profile), their chromatograms might differ, suggesting that their extractables (or leachables) profile could differ” (2). Posgia et al. (3), reporting on a blinded round-robin study to investigate interlaboratory variations related to compound identification and quantitation and to evaluate whether lab selection impacts test results, noted that significant differences were observed among the test laboratories in compound identification, confidence in the identification, and quantity reported and concluded that the selection of a test laboratory could have a significant impact on a risk assessment. Sawyer (4, 5), reporting on the results of a round-robin exercise to study the extraction conditions used for the biological testing of medical devices, noted that “Lab to lab variability in the number of compounds identified was noticeable” and suggested that “the lack of agreement between chemistry test laboratories calls into question the reliability of chemical extract data to support toxicological risk assessments used in the biological evaluation of medical devices”.
If the impact of this lab-to-lab variation was largely academic, then its analysis and resolution would be largely a matter of scientific debate. However, as extractables and leachables data are the input into a toxicological safety risk assessment, any lab-to-lab variation in an extractables or leachables profile could have a profound effect on the conclusion of the risk assessment, whose ultimate purpose is to establish whether a drug product or medical device is safe for its intended use.
If one accepts the hypothesis that there is lab-to-lab variation in reported extractables/leachables profiles, then the E&L community of practice must accept their responsibility to identify, mitigate, and/or eliminate the variation's root cause(s) and thereby reduce variation. If lab-to-lab differences in analytical processes and procedures drive lab-to-lab variation in reported profiles, then it is logical that removing the differences by standardizing processes and procedures is an appropriate means of addressing this issue.
Unfortunately, standardization of processes and procedures is best accomplished prospectively and not retroactively. Standardization of the chromatographic processes for screening extracts for extractables and drug products for leachables today would have to deal with the significant headwind of resistance to adoption of the standardized processes. That is, it is almost certain that laboratories would hesitate to voluntarily abandon their existing methods, given the capital and intellectual investments that they have made in those methods and the historical data that has been generated by those methods. Moreover, development of the standard methods is both scientifically and practically complex, presuming that the scientific community is both technically mature enough to establish the methods and emotionally mature enough to accept consensus methods. Furthermore, someone or, more likely, some organization, would have to accept the responsibility for mandating the standard and monitoring adherence.
Additionally, advancing technology presents a challenge to standardization, either perceived (as in everybody wants to use the latest toy) or real (as in replacement of the standard method with a method that produces output with improved quality [e.g., more accurate] or practicality [e.g., less expensive]).
Lastly, standardization of process does not necessarily drive consistency in process implementation. In the same way that two cooks working from the same recipe might produce profoundly different deserts, two laboratories working from the same standard might still produce differing extractables or leachables profiles, depending, for example, on the instrumentation used and the skills/training of the analysts.
If the challenges of standardizing extractables and leachables screening methods and processes appear to be insurmountable, then perhaps the approach of standardizing the output can be considered. Rather than standardizing the process by which an output is generated, one standardizes the output by establishing a minimum quality standard for the output. Methods that are able to meet the minimum quality standards are said to be standardized and likely will produce comparable lab-to-lab data, regardless of any methodological differences between labs.
Minimum quality standards reflect both qualification of a method (the method's ability to produce the necessary output) and qualification of the output (i.e., what type of output is established as being acceptable for toxicological risk assessment purposes). Relevant minimum quality standards can be applied to the three major objectives of screening, each of which is addressed as follows:
Detecting and reporting a compound,
Identifying a compound, and
Quantifying a compound.
Detecting and Reporting a Compound
Before a compound can be identified and quantified, it must be detected. Once a compound has been detected, the decision whether to report the compound, potentially triggering the actions of identification and quantitation, must be made and justified.
The “detection power” of an analytical method is measured in two dimensions, the number and chemical diversity of compounds that produce a response, that is, the number of peaks produced (breadth and coverage) and the magnitude of the response produced, that is, the size of the produced peaks (sensitivity).
Breadth and Coverage of the Method.
As a chromatographic separation method targets compounds with specific physicochemical properties (e.g., largely semivolatile compounds for GC, semivolatile and largely nonvolatile compounds for LC), a method's breadth or scope is defined as the number of compounds that produce a recognizable response, which is linked to the responding compounds' key physicochemical characteristics. Chromatographic screening methods applied to extractables and leachables are often described as being broad-scope or having a considerable breadth, as it is desired that the methods produce an adequate response for a large number of chemically diverse compounds.
The term coverage refers to the ability of the chromatographic method to respond to all possible organic extractables/leachables and thus is related to breadth. However, whereas breadth is related to the number of compounds producing a viable analytical response, coverage is related to the percent of compounds in the entire population of potential extractables/leachables that produce a viable analytical response. Logically, a method with a greater breadth (greater number of compounds producing a useful response) will have a greater coverage.
Although the following discussion specifically addresses breadth, it is equally applicable to coverage.
One measure of a method's breadth is the method's list of detectable compounds. As the method is used, the method's list of detected compounds grows and a review of the list allows the method's breadth to be established (i.e., “good for C1–C10 alkanes, not so good for branched fatty acids”). Typically, the list grows as the method is applied to specific projects and likely the contents of the list will vary somewhat from lab to lab depending on the products and projects supported by a lab. Thus, for example, use of the same method by two labs, one specializing in medical device testing and the other specializing in packaged drug product testing, is likely to result in breadths that consist of slightly different specific compounds but similar compound classes. As the list grows, likely the method's breadth expands somewhat and that breadth can be more precisely defined.
As an alternative or supplement to this “making a list and checking it twice” approach to defining breadth, breadth can be demonstrated by a method's ability to respond to intentionally chosen marker compounds. Thus, for example, compounds that elute early and late (so-called anchor compounds) can be used to establish the “boundaries” of the method whereas marker compounds of a particular functionality (or of known significance) can establish the ability of the method to respond to necessary compounds or compound classes. For example, alternative markers chosen to reflect functionalities might include (as appropriate) acids, alcohols, aldehydes, ketones, siloxanes, and so forth, whereas supplementary markers chosen to reflect significance might include (but are not limited to) so-called “bad actors”, such as di-(2-ethylhexyl) phthalate (DEHP) and bisphenol A (BPA) as compounds of known toxicological concern and bis(2,4-ditert-butylphenyl)phosphate (bDtBPP) as a compound known to reduce yield in biomanufacturing (6).
Comparing a method's experimentally determined breadth, checked by testing a set of marker compounds either during an initial method qualification or at the time of use via system suitability testing, to a list of “must have” compounds (prepared by expert consensus) serves as a means of standardizing methods. Methods from various labs that produce an acceptable response for each member of the set of critical compounds are said to be standardized with respect to breadth.
Sensitivity of the Method.
The Analytical Evaluation Threshold (AET) is that concentration of an organic extractable or leachable at which the compound is likely enough to be unsafe that it requires formal safety assessment. The AET is typically set at the Safety Concern Threshold (SCT), the latter being the concentration of an organic extractable or leachable at which the compound is likely enough to be unsafe that it requires formal safety assessment. Compounds at levels at or above the AET are thus reported for safety assessment. It is intuitively obvious that the sensitivity of a chromatographic screening method must be sufficient that extractables can be detected at least at the level of the AET, if not lower, and thus the requirement that the limit of detection (LoD) ≤ AET is appropriate for all testing laboratories.
Let us envision the desirable situation in which all labs calculate the AET in the same manner (e.g., they all use the same uncertainty factor [UF] to account for response factor variation) and thus all labs are using the same value for the AET. Assuming that all labs meet the requirement that LoD ≤ AET, then at least the number of reported compounds at or above the AET should be highly similar from lab to lab because all labs are able to meet the same reporting threshold. However, as the labs may have different LoDs, it is reasonable to expect that they may have detected a different number of compounds below the AET. If the labs choose to report these compounds, then there is a clear opportunity for the two labs to have two different lists of extractables.
This situation is readily and easily managed by asking labs to report all compounds at or above the AET in a different manner (or in a different place in the report) than compounds below the AET but above the LoD. In so doing, the lab-to-lab extractables profiles will be more similar at or above the AET, dispelling the perception that lab-to-lab differences in extractables profiles are rampant in the industry. Although there may be greater lab-to-lab difference in the profile below the AET, such differences are largely academic from the perspective of adversely affecting the rigor and adequacy of the safety assessment, as compounds below the AET are not safety assessed.
Another aspect to consider about the AET is how it is applied. For example, the AET response threshold applied to all compounds can be established based on the response of a single compound, the mean response of all compounds, or a customized response for groups of compounds (a different “drawing of the AET line” for different compound classes, i.e., one line for phthalates, another line for siloxanes, and so forth). Although simplicity supports the “one size fits all approach”, good science suggests that “customizing sizing” is most rigorous. As a compromise between practicality and science, this author suggests that using the mean response to anchor the AET is a good first step toward standardization.
In a certain way, the aspect of sensitivity for the screening method is a refinement of the aspect of breadth. Whereas breadth reflects the ability to produce a detectable response, sensitivity requires that the magnitude of the produced response be above a fixed value. Given this similarity, the approach taken to establish sensitivity is essentially the same as the approach taken to assess breadth and, in fact, it is most efficient if the breadth and sensitivity are assessed by the same data produced by the same experiment. However, breadth is an invariant characteristic of an analytical method whereas sensitivity is a situational characteristic of an analytical method, as the AET can vary from situation to situation. Thus, it is possible that two labs could have methods with equal breadths but an unequal ability to meet the sensitivity requirement.
The preceding discussion works well in the situation in which the AET is achievable by all testing laboratories. However, one can envision situations in which the AET is so low that it is an insurmountable challenge for one or more (or perhaps all) labs to meet the expectation that LoD ≤ AET. Invariably and unavoidably, this differing ability to “go low” will lead to lab-to-lab variation in the reported extractables/leachables profiles.
Mathematical Toxicological Safety Assessment Simplified
It is beyond the scope of this correspondence to address the science and practice of toxicological safety risk assessment, especially regarding the proper means of establishing a compound's tolerable daily intake (TDI). For further insights into the complexities involved in establishing a properly rigorous and conservative TDI, the reader is referred to references 7⇓⇓–10. These complexities notwithstanding, a high-level description of the mathematical component of safety risk assessment is relevant when considering how the risk assessment can account for analytical variation in compound identification and quantitation.
Greatly oversimplified, the toxicological assessment involves comparing a patient's daily exposure to a leachable (DE) to an exposure level that is considered to be safe, such as the TDI. A margin of safety (MoS) can be calculated as the ratio of DE to TDI via eq 1:
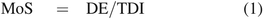
If the DE is greater than the TDI (MoS ≥ 1), the compound of interest is generally assessed as having a negligible adverse effect on patient health and safety.
Considering the terms in eq 1, DE is calculated from the level of a leachable in a drug product (CL) and the drug product's maximum daily dose volume (MDDV). Thus, the DE can be adjusted to account for the uncertainty in CL by adding a UF to the DE calculation, for example, eq 2:
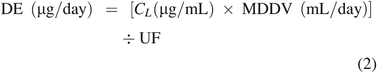
Considering the TDI, TDI is established based on the identity of the compound of interest, as identity is the link between the compound and its toxicological data. Thus, the TDI can be adjusted to account for uncertainty in the leachable's identity by adding a UF to the TDI calculation per eq 3:
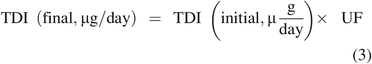
The net result of the UF adjustment to either DE or TDI (or both) is a smaller value for the MoS and therefore a more conservative estimation of a compound's potential toxic effect.
Identifying a Compound
It is relatively accurate to observe that everybody follows the same process for identifying extractables at a high level. This is true as identifications are typically secured in one of three ways:
Matching of compound-related information (mass spectrum, retention time) to libraries and/or databases of compiled information,
Expert interpretation of the analytical data (structure elucidation from an interpreted mass spectrum), or
Interpretation of related information (composition, similarities between extractables).
However, it is in the details where individual practices differ. Considering matching, for example, individual practitioners may have different criteria as to what constitutes an acceptable match.
Thus, it can be a difficult task to develop standardized practices for establishing the identity of a compound that produces a chromatographic peak (presumably with mass spectrometric detection).
Alternatively, standardization can be driven by requiring that identities reported for safety assessment meet a certain standard of science. To accomplish this purpose, I note that identifications have already been classified with respect to their scientific rigor, see, for example, Table I (9). Thus, one notes that a confident identity supported by two corroborating pieces of evidence (e.g., spectral matching and structure elucidation) is more scientifically rigorous than is a tentative identity supported by a single piece of evidence (e.g., spectral matching or structure elucidation). It follows, then, that a safety assessment performed on a confidently identified compound is likely to be more correct in establishing the compound's patient safety impact than an assessment performed on a tentative identity.
Identification Categories. The Bolded Category is the Minimum Reporting Requirement
As Identification Categories have already been created and are routinely reported (9), the challenge for this Correspondence is simply establishing that level of identification that is sufficiently rigorous to serve as a standardized basis for safety assessment. To this end, it is the opinion of this author that all safety assessments should be based on at least confidently identified compounds, where a confident identification is based on two largely independent pieces of collaborating evidence.
This opinion notwithstanding, it is naïve to expect that confident identities can be secured for each and every extractable in each and every circumstance. This means that saying “all IDs must be confident” without allowing for exceptions ignores the practical realities of E&L analysis and thus is not actionable guidance. There are many circumstances in which the collection of the two independent pieces of collaborating information required for a confident identification is so practically and scientifically challenging as to be nearly impossible. It seems clear to this author that rather than forbidding the use of lower confidence identities in safety assessment, a more actionable approach would be to require that there be a consequence for using lower confidence identities. That is, a lower confidence identity could be used as the basis of a safety assessment, but only when reasonably rigorous options for obtaining a more confident ID have been exhausted, such as spectral matching, structure elucidation, and the collection of confirmatory information. However, in so doing, one must account for the additional uncertainty when lower confidence identities are used, perhaps by adjusting the DE to a lower value. Specifically, when one uses a partial or tentative identity, a UF, assigned a value of no greater than 2, is used in the calculation of the DE.
Now it could be suggested that a UF should be higher for a partial identity than for a tentative identity, as the partial identity is more uncertain. However, this true statement is typically addressed during the toxicological safety assessment. That is, if a partially identified compound is safety assessed, it is typically assessed by either read-across to a surrogate or use of a Threshold of Toxicological Concern (TTC), for example the mutagenicity TTC per ICH M7, as the TDI. In the case of read-across, an additional UF is usually included in the TDI determination. In the case of use of a TTC as the TDI, it is noted that by its nature, the TTC is highly conservative and thus a TDI derived from a TTC is highly conservative. In these ways, the safety assessment of a partially identified compound is already sufficiently conservative that using a larger UF to account for the increased uncertainty of a partial identity is, in a certain respect, akin to “double dipping”.
Quantifying a Compound
The aspect of quantifying a leachable can be similarly addressed in terms of accounting for uncertainty in the leachable's calculated concentration in the safety assessment. As was the case for identification, quantitation categories have already been developed and are in common use (see Table II) (11). As quantitation categories have already been created and are routinely used in reporting, the challenge for this Correspondence becomes simply establishing accuracy requirements for each category and establishing categories that are a suitable basis for safety risk assessment.
Accuracy Scale for Quantitation. The Bolded Category is the Minimum Reporting Requirement
Assigning required levels of accuracy to the quantitation categories is accomplished in Table II. Given these accuracy levels, this author suggests that a semi-quantitative determination of concentration, typified by an accuracy of “plus or minus a factor of 2” is adequate for safety risk assessment of data produced in screening analyses. A higher level of quantitation is typically required in target analysis.
As was the case with identification, it is naïve to require that all safety assessments be based on semi-quantitative data (or better), as there are likely circumstances in which an estimated concentration is as good as one can get. Rather than precluding such estimated concentrations from safety risk assessments, this author recommends that a UF =2 be applied to the determination of the DE if the DE is based on an estimated concentration (see eq 2). As was the case for identity previously, use of the UF in quantitation produces a smaller MoS and thus a more conservative safety risk assessment.
It is reasonable to consider which of the quantitation methods that can be employed in E&L screening can meet the accuracy criteria, not for individual compounds but for what is likely a suite of compounds whose concentrations must be determined. It is almost a certainty that the use of individual standard curves for each analyte will meet the requirement for a quantitative method. It is possible that the use of analyte-specific relative response factors (collected in a database), which are essentially one point calibration curves, will meet the requirement for a quantitative method and is highly likely to meet the requirement for a semi-quantitative measurement. It is this author's opinion that the matching of an analyte to a surrogate standard, whether by retention time or structure, will not unilaterally result in a quantitative result, as it requires great skill and technical knowledge to link the analyte with an appropriate surrogate. It is highly unlikely that use of an internal standard as a single surrogate standard will produce a quantitative result. Whether the result is semi-quantitative or estimated depends on the variation in the response across analytes and concentrations as well as the laboratory's skill at choosing an acceptable surrogate. For example, consider the case of GC/MS. Several authors have suggested that response factor variation in GC/MS is generally “a factor of 2 either high or low” (1, 12⇓–14) when the internal standard is chosen at the mean or median response of a response factor database. In this case, the accuracy requirement of 50% to 200% can be met and the method can be established to be semi-quantitative. However, improper choice of the surrogate (i.e., picking an internal standard above or below the median), will produce accuracies that are either biased high or low and that will fall outside of the range for semiquantitative, meaning the method produces only an estimated result (15).
The situation is much less promising for LC/MS where response factor variation is much greater than our “factor of 2 either way” (1, 13, 16). Thus, LC/MS quantitation with a surrogate or internal standard will likely produce a reported result that is no better than an estimated result, meaning that LC/MS concentrations based on a single surrogate standard are inherently estimates. Thus, safety risk assessment based on LC/MS concentrations must take into account the lower accuracy of such concentrations (e.g., the use of UF = 2). However, augmenting LC/MS data with supporting data, such as that obtained via the use of multiple detection methods, may provide a means of producing semi-quantitative data via LC methodology. This is the case, as use of multiple detection methods increases the likelihood that semi-quantitative results will be achieved by at least one of the detection methods.
Conclusion
Standardization of chromatographic screening methods for organic extractables and leachables will certainly improve the degree to which the reported extractables (or leachables) profile agree across testing laboratories. Although standardization can be achieved by all laboratories adopting the same analytical procedures, processes, and methodologies, there are many headwinds that make it unlikely that such standardization will occur. Thus, this Correspondence proposes that a certain degree of standardization can be achieved by establishing requirements and specifications for the screening method's output (i.e, the reported extractables or leachables profile). Supporting this proposition, this Correspondence established the following performance expectations:
To address a method's Detectability:
Breadth is established by verifying that the method produces an acceptable response to all members of a selected, justified, and generally accepted test mixture containing known extractables and/or leachables.
Coverage is established by the laboratories' determination and use of the AET. The requirement with respect to the method providing acceptable coverage is that the AET be linked to an internal or surrogate standard whose response factor is equal to the median of the response factors for a relevant population of E&L compounds and that an AET so linked to the proper internal standard be adjusted via a justified response factor UF, which is generally between 2 and 5. The combination of the linkage and adjustment produces a final AET.
Sensitivity is established by noting that the method's limit of detection for all compounds in the justified test mixture (see Breadth) be less than or equal to the final AET (see Coverage).
To address a method's ability to support compound identification, the requirement is that any and all identities reported for the purpose of patient safety risk assessment must achieve the Confident level, meaning that the identity is supported by two independent but corroborating items of evidence. If tentative or partial identities are used in toxicological risk assessment, the added uncertainty must be accounted for in the safety risk assessment; for example, by applying a UF of no more than 2 to the tolerable daily intake value.
To address a method's quantitative ability, the requirement for screening chromatographic methods is that all concentrations reported for the purpose of safety risk assessment must be semi-quantitative, herein defined as a method that has been demonstrated to produce acceptable accuracy (e.g., 50%–200%) for all members of a selected and justified test mixture. If this criterion is not met and an estimated concentration is used to establish the patient daily exposure, the added uncertainty must be accounted for in the safety risk assessment; for example, by applying a UF of no more than 2 to the daily exposure.
An important aspect of this proposal is its alternative approaches. Although it is highly desirable that standardization be achieved by meeting expectations for output, it is recognized that there may be circumstances in which meeting the specifications are either not practically possible or unnecessary. Considering the practically possible, it is unrealistic and frankly unfair to require semi-quantitation and confident identities with no recourse. There will be circumstances in which the level of effort required to elevate an estimated concentration to semi-quantitation and/or a tentative identification to confident is beyond reasonable expectations. Rather than inform the unlucky sponsor in this situation that “these are the rules and you meet them or else”, the approach proposed herein allows for an alternative tactic but exacts a price for using that tactic; that is, reduction of the DE or increase in the TDI via application of a UF. Thus, uncertainty in the measurement is taken into account by use of UFs.
Even if likely an identification, quantitation, or both can be elevated with a reasonable effort, it may still be the case that the effort is unnecessary. For example, consider a situation in which a tentative identity and an estimated concentration are the points of departure for a toxicological safety risk assessment, embodied in the MoS. Furthermore, the situation is such that the calculated MoS of the tentatively identified, estimated concentration extractable is large, for example 100. In this case, application of UFs for both identification and quantitation would reduce the calculated MoS to 25, which for all practical purposes still establishes that the extractable in question is highly unlikely to adversely affect patient safety. In this situation, a definitive conclusion about safety impact can be drawn when the UF correction is used and thus it may be easier to use the UF correction than to secure the better (higher quality and certainty) identity and concentration.
The concept of a test mixture has been mentioned several times in this Correspondence. Perhaps it is a deficiency of this Correspondence that the test mixture has not been specified. Failing to do so is a simple matter of competence and collaboration. It is likely the case that the least qualified person to specify the test mixture is someone, such as myself, who has not sat in front of a chromatographic instrument as an operator for almost a decade. It is also likely that there will be some degree of professional debate as individual experts and/or individual organizations have their own opinions in terms of which compounds (and how many compounds) make up an achievable but challenging test mixture. No, establishing the test mixture is a team, not an individual, sport and I hope that this Correspondence is a catalyst for the industry to embrace this challenge and get to work.
Moreover, I note that standardization is the only viable solution to the issue of interlab and intralab variation in reported extractables and leachables profiles. If it is to be expected that extractables and leachables profiles be reproducible within a lab and between labs, then standardization is the unavoidable price of admission. This ticket price can either be paid up front, by adopting standard methods, or at exit by requiring standardized performance. Considering the latter approach, which is the focus of this Correspondence, establishing that an analytical process produces output of the requisite quality is otherwise known as method qualification, supported at time of use by system suitability testing.
In closing, I note and freely admit that although the issues addressed in this Correspondence are real, any and all proposed solutions are just that, proposals, some of which may be more solid and sustainable than others. If the end result of this Correspondence is that it facilitates the development of adopted consensus best practices (and then is largely forgotten), then the Correspondence has exceeded its author's expectations.
Conflict of Interest Declaration
The author has no conflicts of interest to disclose; however, Dr. Jenke notes his affiliation with a contract research organization (CRO) that provides extractables and leachables services to the pharmaceutical and medical device industries.
- © PDA, Inc. 2023



