
Abstract
To address patient safety, a drug product is chromatographically screened for organic leachables. Similarly, medical device and packaging system extracts are chromatographically screened for organic extractables as probable leachables. To protect patient health, the screening methods must respond to all potentially unsafe substances. To be efficient, analytes determined to be below the toxicologically relevant threshold are removed from consideration before the subsequent analytical tasks of identification and quantitation are performed. The analytical evaluation threshold (AET) was established for use as a toxicologically relevant threshold to differentiate between chromatographic peaks that are unlikely to be unsafe (and thus do not need safety assessment) and those that are possibly unsafe (and thus require safety assessment). In practice, the instrumental response associated with the AET is determined using surrogate standards. It is then assumed that the response strength for an unknown extractable is equivalent to that for the surrogate standard at the AET concentration (i.e., relative response factor = 1). It is an unfortunate reality that response factors vary for different compounds on nearly all detector systems. This complicates the application of the AET and can result in a failure to include potentially toxicologically relevant compounds in the identification phase of the analysis. To ensure protection, an uncertainty factor was built into the AET equation that adjusts the AET downward to address response variation. Although this mechanism does increase the protectiveness of the AET, it assumes that the available methodology and instrumentation is sufficiently sensitive to reach the new lowered AET value. However, in some cases, the response variation is so great and the resulting uncertainty factor so large that the revised AET is below the achievable sensitivity specifications of even state-of-the art, expertly operated instrumental technologies. The only option then remaining is to concentrate the samples, which can result in adverse effects on analysis quality—counteracting the perceived benefit of lowering the AET. This article demonstrates how an analytical strategy based on methods with multiple complementary and orthogonal detection techniques (a multidetector approach) mitigates the problem of response factor variation and thus eliminates the need for large uncertainty factors and the resulting lower AET values. The primary concept is that all analytes only need to be effectively detected by at least one of the combination of detectors applied, and it is this effective technique (combination of all detectors and chromatographic methods utilized) that is used to perform the AET assessment.
- Extractables
- Leachables
- Analytical evaluation threshold
- AET
- Uncertainty factor
- Response factor variation
- Multidetector approach
Introduction: The Analytical Evaluation Threshold
Leachables in drug products are foreign impurities derived from the drug product’s packaging and manufacturing systems. These substances are transferred to a patient as the patient is clinically treated with the drug product. Leachables in medical devices are substances present in the device that transfer from the device to the patient during the device’s use with the patient (1⇓–3). In either circumstance, the effect of the leachables on patient health and safety is of paramount concern.
The purpose of an extractables/leachables study is to establish either those extractables that could leach by performing a controlled extraction study on the medical device or packaging and/or manufacturing system or to establish those leachables that have leached by performing a migration study. In either case, the extract, the drug product (DP), or the contact medium between a medical device and the patient is screened for organic extractables or leachables using chromatographic methods to discover, identify, and quantify these substances.
An important consideration in screening for extractables and leachables is “what limit of detection is required?”, or alternatively “what is the concentration of an extractable which is so low that likely the substance is safe regardless of its identity?” Substances that are below this limit do not need to be identified to establish that they are safe, thereby reducing the analytical burden of identification. This is highly significant, because in certain circumstances (such as aggressive extraction of a chemically complex medical device or container closure system [CCS]), the number of peaks that need to be reported is large and the difficulty of securing identities for all peaks is considerable. In such circumstances, it may be challenging, if not impossible, to produce credible identifications for all substances detected at trace levels.
To this end, the concept of the analytical evaluation threshold (AET) was developed by an Extractables and Leachables Working Group of the Product Quality Research Institute (PQRI) to facilitate the toxicological risk assessment of extractables and leachables (4). This concept was then further extended to medical devices in a recent update to the ISO 10,993-18 standard as shown in eq 1 (5):
 (1)
(1)
In which DBT is a dose-based threshold such as the threshold of toxicological concern, A is the number of devices extracted, B is the extract volume, C is the maximum number/mass of devices used per patient, D is the dilution or concentration factor, and UF is an uncertainty factor applied to account for response factor (RF) variation.
The AET establishes that level at and above which organic extractables or leachables must be reported for toxicological risk assessment (6). In order for a reported leachable to be toxicologically risk assessed, the analytical method must discover the leachable and provide information that leads to the leachable’s identity and concentration in the drug product or extract. This makes the AET a “protective” threshold, because the identification and risk assessment of all compounds above the AET is performed to protect patient health and safety.
The application of the AET in chromatographic screening is illustrated in Figure 1. Once the AET is calculated, a surrogate standard, sometimes included as an internal standard, must be analyzed at the AET concentration to determine the instrumental threshold. A line at the AET concentration is drawn across the chromatogram using the apex of the surrogate standard. Peaks whose responses are at or above the line are determined to be in the product at a level greater than the AET and must be reported for toxicological risk assessment. Peaks whose responses are below the line are not reported for toxicological risk assessment as they are deemed to be at such a low concentration as to pose no significant risk.
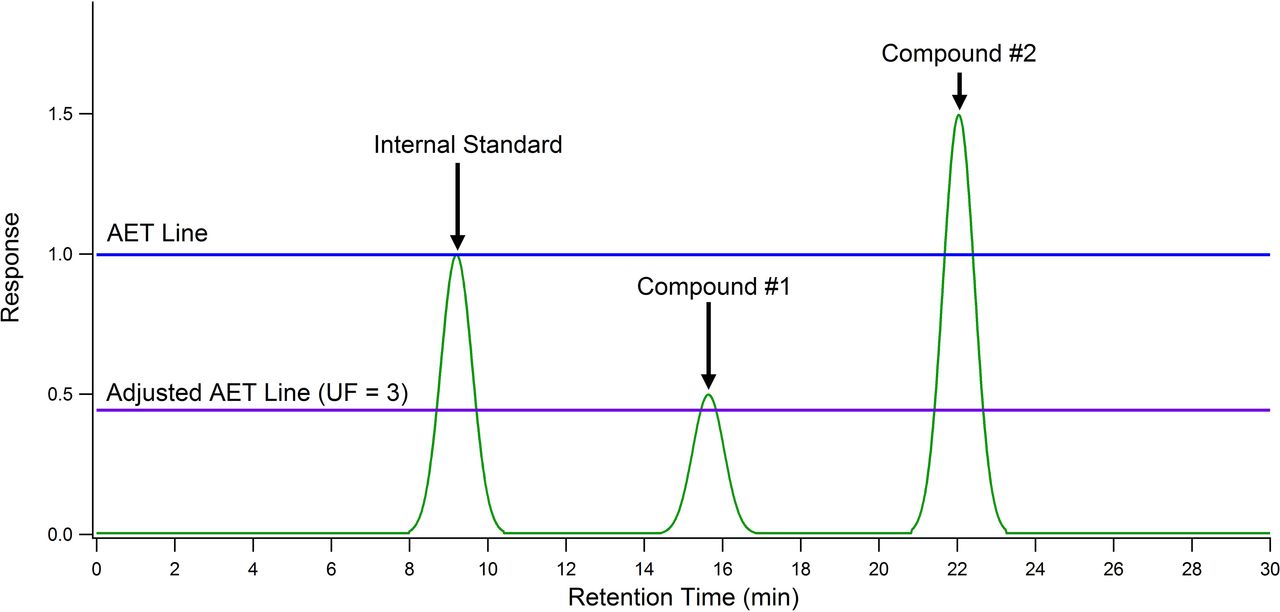
Analytical evaluation threshold (AET): pictorial representation of the AET. An internal standard has been added to the extract at a concentration equal to the AET. When the extract is analyzed, a line at the apex of the internal standard peak is drawn horizontally across the chromatogram. Peaks whose responses are above the line must be reported for toxicological risk assessment. Peaks whose responses are below the line need not be reported for toxicological risk assessment, as they are deemed to have a negligible adverse effect on patient safety. Application of the AET in this manner is based on the assumption that the internal standard and the analytes have the same response factor. Assuming all three compounds are at an equal concentration (AET concentration), an “AET gap” is caused by differing response factors. This gap is corrected by using an uncertainty factor that takes into account response variation between analytes and internal standards. This has the effect of moving the AET line lower in the chromatogram and results in compound #1 being successfully “captured” by the AET assessment.
In practice, chromatographic responses are rarely quantified using peak heights and thus the AET is typically applied as a peak area threshold. Additionally, it may not be practical to spike the drug product, CCS, or medical device extract with an internal standard at exactly the AET concentration, in which case, the AET “line” is drawn at some height of the internal standard peak equivalent to the AET. Lastly, it may also be advantageous to determine the threshold using the average response for a group of external standards for reasons that will be described later.
Application of the AET implicitly presumes that the RF (see eq 2) for every organic leachable and surrogate standard candidate is the same; that is, equal concentrations of leachables and surrogate standards produce the same magnitude of response. Unfortunately, this presumption is not accurate for most commonly employed chromatographic detectors. Thus, the simplistic application of the AET as described to this point is prone to error, where the position of strongly responding analytes is exaggerated versus the AET and the position of weakly responding analytes is understated versus the AET. This can result in false negatives and exclusion of compounds from the identification and quantitation phases that are in fact above the AET level. It can also result in false positives such that compounds below the threshold are toxicologically risk assessed even though this effort is not warranted.
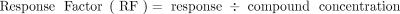 (2)
(2)
If the root cause of the AET error is differing RFs among analytes, then one way to address the error is to adjust the AET downward to account for RF variation. This is the purpose of the uncertainty factor (UF) in eqs 1 and 3. The magnitude of the variation that must be accounted for using the UF is dependent on the magnitude of the RF variation between analytes and surrogate standards. Mathematically, such a correction to the AET is shown in eq 3, which is a simplified version of eq 1:
 (3)
(3)
The UF has previously been related to the RF distribution for all leachables and extractables analyzed by the method using eq 4 (7, 8). Mathematically this is typically accomplished by calculating the relative standard deviation (RSD) from an RF database created using the specific analytical method applied and extractables selected so as to be representative of the variety of the compounds that are expected to be analyzed with the method. By adjusting the AET downward via the UF, analytes with RFs lower than that of the surrogate standard now have responses that exceed the revised AET and thus are properly reported for toxicological risk assessment. An example of this can be seen in Figure 1, where a UF of 3 was applied to adjust the AET line downward to properly “capture” a previously unreported compound.
 (4)
(4)
Although eqs 3 and 4 are mathematically uncomplicated, there are issues in establishing a useful and acceptable UF. Firstly, determination of the RSD of the distribution for all leachables and extractables intended to be analyzed by the method presumes that the analyst knows which extractables and leachables will need to be identified. This presumption of the relevant potential extractables and leachables is generally based on a review of the chemistry of the materials of construction and early screening results and can be termed focused screening. Although some information is generally available regarding the chemistry of expected extractables and leachables when well characterized materials of construction are being investigated, examples of unexpected deleterious leachables have been reported, highlighting the weaknesses inherent in a focused screening approach. Examples such as the recall of Tylenol because of 2,4,6-tribromoanisole from wood pallets or the recall of breast implants because of tainted industrial grade silicones with unexpected impurities have shown that harmful leachables are not always predictable based on the chemistry of the medical product under study (9, 18). For this reason, an unbiased universal screening approach is preferred for the safety assessment. This is not to deny the value in using relevant chemical standards but rather to highlight that the assumption that all relevant toxic impurities are predictable has been shown historically to be false. Another complication is limited availability of analytical standards for all extractables and leachables, preventing the determination of the RF values. In the absence of a complete list of extractables and without ready availability of reference standards, an estimate of the RSD of the distribution will have to be substituted for the true RSD. The accuracy of this estimate then depends on the equivalence of the RSD for the surrogate standards used in the focused screening approach and that for the true distribution. This raises questions as to how such an estimate should be confirmed to be representative, especially in the context of a focused screening approach using a limited number of standards. In comparison, approaches intended for universal screening can be continuously evaluated as more extractables and leachables are analyzed with the method providing an ever-improving estimate of the true RSD of the method for the universe of extractables.
Another significant problem is the large amount of RF variation for some detection methods such as liquid chromatography–mass spectrometry (LCMS), which then results in high RSD values (8, 10, 11). Practically speaking, as the RSD increases past 0.8 (a number that is not unusual for LCMS RFs), the value of the UF rapidly increases. To a certain point, this escalation in the UF is more a matter of practicality, as the revised AET becomes smaller and more difficult to achieve as the UF increases. However, at the point where the RSD reaches and exceeds 1, the UF actually loses its physical significance as it produces an undefined result (when the RSD = 1) and then becomes a negative number (as RSD becomes >1). In such cases, an AET cannot be calculated using eq 1. Furthermore, if the analytical method is not sufficiently sensitive to reach the revised AET, then compounds that are potentially unsafe will not be detected and the method is not protective of patient health and safety. It does the analytical chemist little good to have a revised AET that cannot be achieved.
Mass spectrometry detectors are the “de facto workhorse analytical instruments for extractables and leachables (E&L) studies” (12). This is primarily a result of their identification capabilities combined with their generally high sensitivity and wide applicability. Alternative detectors (such as FID, UV, CAD, and so forth) have typically not been emphasized in extractables and leachables work, but examples of their use for drug impurity screening are not uncommon (13⇓⇓–16). One significant advantage of using two detection methods is that now there are two different analytical signals upon which to judge whether the compound is above the AET or not. Although either detector could be used for this assessment, intuitively it is appropriate to base the determination on the best method for the particular analyte. Arguably, that best method is the one that has the least amount of RF variation, as this would result in the smallest UF and the least necessary adjustment of the AET. If this concept makes sense for two detection methods, its value is even greater when more detection techniques are employed. In this case, one has information from multiple different analytical signals upon which to judge (1) whether the compound responsible for the peaks in one or more of these combinations is above the AET, (2) what the concentration of the compound is, and (3) what the identity of the compound is.
The strengths of the multidetector approach are clear. First and foremost, such an approach more comprehensively covers the entire universe of potential extractables and leachables, as an extractable that does not respond to one detector is likely to respond to another so long as their mechanisms of detection are independent. Secondly, the approach produces a wealth of supporting information that informs and enables accurate quantitation and correct identification. The purpose of this publication is to describe a means for evaluation of the AET using a multidetector approach and to demonstrate how it can be used to mitigate the effects of RF variation and thus reduce the need for large UF.
AET Evaluation Using Multiple Orthogonal and Complementary Separation and Detection Methods
When considering the results obtained using a multidetector approach, the first decisions that must be made is “which detector signal should be applied for AET determination?” It is possible to get conflicting information from the various detectors, especially if a revised AET is applied and one or more of the methods has high RF variation. Thus, possibly one (or more) detector would indicate that a compound is “above the AET” whereas other method combinations would say “it is below the AET”, creating the dilemma of “which one is right?” Three approaches could be applied to this situation: (1) majority rules (and hope there is not a tie), (2) the result from the best method for the analyte is taken (RF value closest to that for the internal standard), or (3) if the peak is above the AET in one detector, it is above the AET period. At this early stage in the analysis, identification has not yet been performed and hence there is limited information upon which to gauge which detector system would be optimum for an individual extractable or leachable. For this reason, the third option (if a peak is above the AET in one detector, it is above the AET) seems a necessary approach until more information on the compound’s identity is obtained.
To further illustrate how AET evaluation can be conducted using a multidetector approach and how it can prevent unreported compounds at the AET, consider the following data from a recent publication by the same primary author (10). In this work, a combination of five different detectors were utilized as a part of two chromatographic systems. The first system consisted of a quadrupole time-of-flight liquid chromatography mass spectrometer (QTOF-LCMS) coupled with a charged aerosol detector (CAD) and an ultraviolet-visible detector (UV). This system is denoted as a QTOF-LCMS-UV-CAD. The second system was a dual detection gas chromatography mass spectrometry (GCMS) system using a Polyarc reactor system with flame ionization detection (FID) denoted as a GCMS-FID. The combination of QTOF-LCMS-UV-CAD with GCMS-FID provides five distinct mechanisms of detection (ionizability for LCMS, light absorption for UV, formation of charged particles for CAD, electron ionization for GCMS, and charged ions from combustion for Polyarc FID). More correctly, the combination includes six detection modes, as the LCMS provides data when operated in either the positive or negative ion mode. The AET was independently evaluated for each detector signal by measuring the relative response factor (RRF) value of each extractable on each detector individually and comparing it with the average RRF value for the distribution of extractables on each corresponding detector as determined using a broadly constituted database of extractables (10). A positive response on any one or more of the six detectors (RRF compound > RRF average/UF) resulted in inclusion of that compound in the study. This method of evaluation was based on using a surrogate standard with a response corresponding to the average for all compounds on that detector to establish the AET on each detector, thus removing potential bias from standard selection.
The goal of combining these detectors was to use the synergies between the detection systems (optimum detection approach for a given chemistry as guaged by taking the highest signal observed among the detectors applied for an unknown extractable or leachable) to substantially reduce the probability that any compounds are not reported when present at or above the AET. It was essential that the detectors be selected such that their mechanisms of detection were independent of one another and therefore poor response on any one detector (such as LCMS) could be compensated for using a detector with an enhanced response for that same compound (say UV or CAD). Pertinent statitical analysis results for the RF distributions for the 217 extractables analyzed using this method are shown in Table I.
Statistical Analysis of a Database of Response Factors
Table II presents the RRF values for five example compounds analyzed using the method. The RRF values were obtained by dividing the slope of a calibration curve (RF) for each compound by the slope of the calibration curve for the surrogate/internal standard. If the resulting RRF value was >1 then the compound would be correctly detected during application of the AET without the need for a UF value using that surrogate standard. This was because an RRF > 1 indicated that the response magnitude for that compound would be greater than that for the surrogate standard at equal concentrations. In Table II, the calculated RRF values were then further normalized using the average RF for the RRF distribution of the 217 extractables on each detector. Normalization resulted in a value of 1 being the average response for the distribution. This normalization compensated for bias in the RRF distribution that could occur from the arbitrary selection of any one surrogate/internal standard that had a response that differed substantially from the average. Because the values were normalized, an RRF > 1 signified a strongly responding compound and an RRF <1 a weakly responding compound in comparison with the distribution average. RRF values for each of the five exemplar compounds were developed in this way to support examination of the multidetector approach (five different detectors configured as two instrumental systems [LCMS-UV-CAD and GCMS-FID]). As stated previously, these systems provided six independent RFs for each compound (five detectors plus one additional signal as LCMS has two RRF values owing to positive and negative mode ionization).
Relative Response Factor (RRF) Values for Five Organic Extractables
As an example of how this approach works, consider the RRF values presented in Table II. A cursory examination of the data reveals that for each compound, the RF values differed for each detector. If the surrogate standard used to set the AET is selected such that it is at the average of the distribution, then any compound that shows an RF value ≥1 would be selected for inclusion in the study using a UF of 1. Similarly, if a UF of 2 is applied then any compound with an RRF > 0.5 (1/UF = 1/2 = 0.5) would be selected for inclusioon. The first compound in the table, bis(2-ethylhexyl) phthalate showed a signal on five detectors (GCMS, FID, UV, CAD, and LCMS positive mode). The normalized RRF values were generally near 1 on all detectors, indicating this compound would be easily selected for inclusion when present above the AET using any one of the detection methods with only a modest UF factor (UF < 2). In comparison, the second compound, dibenzyl phosphate, showed a signal on only three of the five detectors. In LCMS positive mode, the RRF value was only 0.134, indicating that a UF of 7.5 would be required to include this compound in the study using only this detector. Fortunately, the RRF values in negative mode LCMS and in CAD were >1, indicating that no UF factor would be required at all to include this compound when present above the AET so long as these detectors were applied. The third compound was 1,3,5-triphenylbenzene. This compound showed signal on only two detectors (UV and CAD). This means that no UF would be protective for this compound using only GCMS and LCMS. However, using UV, this compound would be selected for inclusion when present above the AET even when no UF was applied, as its RRF value on that detector was 1.94.
A consideration of the last two compounds (4-sulfamoylbenzoic acid and 5-amino-1-pentanol) shows how the multidetector approach makes detection easier for these acidic and basic compounds. Acids and bases are classes of compounds that have often been noted to have poor RFs and are therefore difficult to capture at the AET in MS only methods. Notice how for 4-sulfamoylbenzoic acid, the UV and CAD both provided stronger responses than the MS detectors for this otherwise poorly responding compound. GCMS showed no response for this compound because of its limited volatility whereas LCMS showed a signal only in negative mode with a weak RRF of 0.348. This compound would require a UF of 3 for appropriate detection using only GCMS and LCMS. In contrast, the UV RRF was 2.30, making this compound easily detectable with no need for a UF. Lastly, the basic compound 5-amino-1-pentanol showed very weak response by both LCMS positive mode (0.126) and GCMS (0.095) and thus would require a UF of 8 to be detected when above the AET using only LCMS and GCMS. Fortunately, this compound gave a strong response by CAD (RRF of 2.60) and a reasonable RRF in FID (0.663). This indicates that no UF is required for inclusion of this compound when CAD is applied and a UF of 2 would assure inclusion using FID.
The previous examples made it clear that using a multidetector apporach allows reliable, positive detection above the AET so long as each compound responds strongly on at least one detector (with an RRF equal to or greater than the RRF of the surrogate standard). It is further noted in regards to the UF value that inclusion of a compound in the study will occur whenever the following criterion is met on at least one detector:
 (5)
(5)
To more fully investigate the potential of the multidetector approach to prevent unreported compounds at the AET, an examination of the data for the 217 extractables previously reported was performed (10). Figure 2 shows how many compounds were reported as being above the AET level for the multidetector approach as compared with using only LCMS and GCMS for different UF values. In this plot, the circles and triangles represent the maximum RRF value for an individual compound using the strongest response from among the combination of detectors utilized. The triangles represent the highest RRF using only GCMS and LCMS while the circles represent the highest RRF from the combination of QTOF-LCMS-UV-CAD and GCMS-FID. The analysis demonstrated that the multidetector approach using a UF of 2 resulted in positive detection above the AET for 94% of the analytes. This level of inclusivity was shown to be equivalent (within 1%) to using a UF of 10 when applying only LCMS and GCMS (95% inclusion). This was a strong demonstration of the effectiveness of the multidetector approach for mitigating the need for impractically large values of UF resulting in revised AETs that potentially exceed the sensitivity of even the most modern instrumentation. It accomplishes this with a reduced reliance on sample concentration and work up, mitigating the potential for compound loss, and is thus preferable. Furthermore, it eliminates wasted effort in identifying, quantifying, and risk assessing the excess number of compounds that arise from needlessly low AETs. Finally, as shown previously, some compounds provided no response on MS detectors (e.g., 1,3,5-triphenylbenzene) and thus no UF was protective, whereas the multidetector strategy provided alternative means to overcome this limitation.
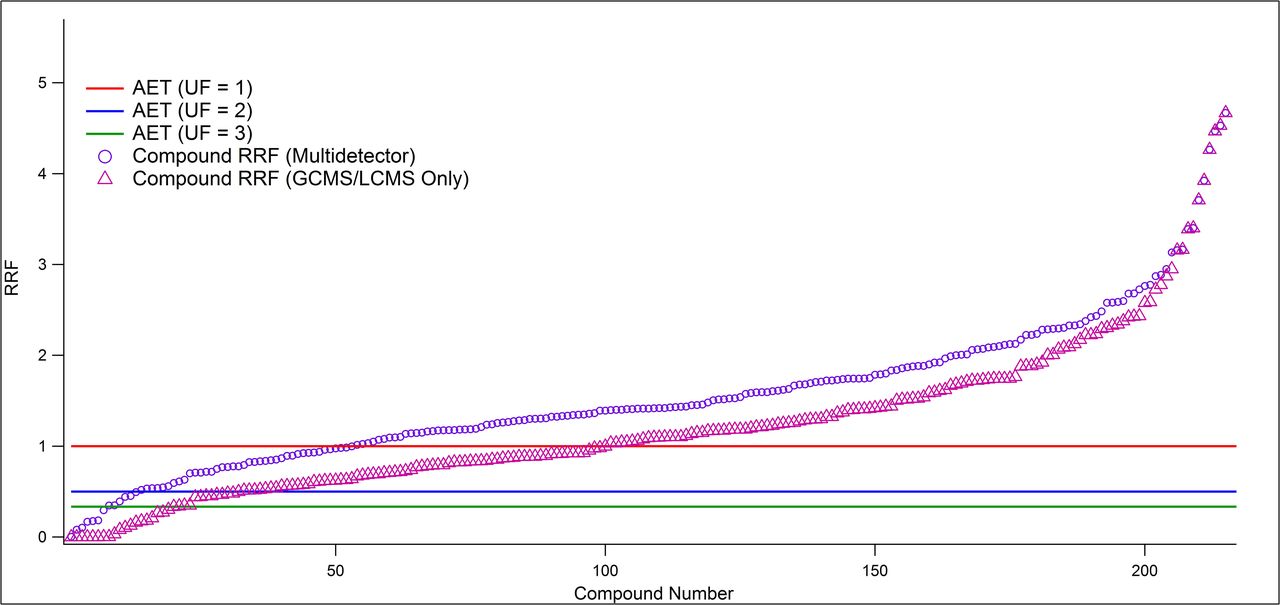
Compound response (relative response factor or RRF) versus analytical evaluation threshold (AET) for different detector configurations. The multidetector approach for mitigating nonreporting. The plot shows the maximum RRF values for liquid chromatography mass spectrometry (LCMS) and gas chromatography mass spectrometry (GCMS) only (triangles) or the maximum RRF value for the multidetector approach (LCMS–ultraviolet-visible detector–charged aerosol detector [LCMS-UV-CAD] and GCMS–flame ionization detection [GCMS-FID]) shown as circles. Normalized RRF values for each compound are presented such that an RRF > 1 indicates a compound would be assessed as above AET at uncertainty factor (UF) = 1 using an internal standard of an average response. As more detectors are added to the analytical strategy, the analytical strategy becomes more protective in terms of ensuring that compounds at a level above the AET are flagged. The location of the AET threshold for a UF of 2 or 3 is also indicated.
Table III compares the protectiveness of the multidetector approach for different UF values ranging from 1 to 4 with different detector combinations. The data indicated that a UF factor was needed in all cases but that smaller UF values were needed to reach the same degree of protectiveness when more detectors were applied. The data also suggested that the magnitude of the number of additional compounds captured decreased as the UF factor became large (less benefit for an equivalent increase in UF). As an example of this, consider what happens when using only LCMS and GCMS as we increase the UF from 1 to 4. The percentage of compounds captured for each increment of the UF is 31%, 6%, and 2% respectively. These diminishing returns are a reflection of the fact that many compounds have already been captured, but also that the detectors utilized may not be fundamentally capable of detecting the remaining compounds. The combination of LCMS and GCMS was found to provide coverage for 54% (UF of 1) to 95% (UF of 10) of the compounds. A nearly equal level of coverage (94%) could be obtained using the multidetector strategy with a UF of only 2. This shows that an equal percent of coverage can be obtained by applying a higher UF value or by adding orthogonal detectors up to about 95% coverage. Beyond this point, the selectivity of the detector systems becomes very significant. Considering the addition of just one orthogonal detector (UV or CAD) to the standard GCMS and LCMS combination, it can be seen that 90% coverage can be obtained at a UF of 2. This compares with 85% for LCMS and GCMS alone at an equal UF value. Thus, adding the CAD or UV provides an increase of 5% coverage at a UF of 2, which is nearly equal in benefit to an increment in the UF from 2 to 3 for LCMS and GCMS alone (6% increase in coverage). Including both CAD and UV increases the coverage by 8% at a UF of 2. This analysis suggested that a balance exists for most detector configurations in which a suitable UF can be identified to provide a reasonable level of coverage, and that this balance favors lower UF as the number of detectors is increased. Finally, it should also be noted that some compounds will not respond on a given detector system and thus although it may be possible to obtain reasonable levels of coverage using high UF values, this approach has definite limitations. As an example, consider that for a UF of 10, 95% coverage is obtained with only LCMS and GCMS whereas 97% coverage was achieved at a UF of 3 for the full multidetector approach. Higher coverage was seen at a lower UF because of the stronger response strength for compounds on one detector system as compared to another. No UF will allow for an accurate assessment of the AET if a compound does not give a response on the detector systems applied
Percentage of Compounds Correctly Reported as at or above the Analytical Evaluation Thresholda
Answers to Some Potential Critiques
One critique of this approach could be that it may result in reporting of additional compounds that are actually present at a concentration below the AET because of their having high RFs. Although this does not pose a negative safety risk, it does increase the work required to complete a safety assessment and thus potentially increases time and cost with no commenserate benefit. Although this possibility does exist, this negative consequence is not unique to a multidetector strategy but can also occur when using large UF values with only the standard LCMS and GCMS detectors. To prevent excess reporting, it is recommended that a two-step approach be applied to determining which compounds are above the AET. The initial AET assessment occurs before identification such that all compounds with a response greater than the AET on any detector are included. Inclusion at this initial stage only indicates that a compound should be identified and its actual concentration further confirmed. Once the identity is known and an appropriate surrogate standard is selected, a second evaluation is performed for each compound to determine if its concentration is actually above the AET. This second assessment is conducted after compound identification and is therefore based on the best available quantitation data.
A second potential critique suggests that it is inappropriate to use UF values selected based on the combination of methods applied and that UFs must be developed solely based on the RF results for each individual detector without considering the combined results for all detectors. This critique tends to be presented in the context of using different chromatographic methods (GC and LC) for RF compensation because they have historically been described as useful for compounds of different volatility (GC for volatiles and LC for nonvolatiles). It is our opinion that this critique is based on a misunderstanding of how a multidetector strategy is applied to AET evaluation. To better illustrate the multidetector approach, consider again the data provided in Table II. For this demonstration, we will consider the compounds presented as a database of relevant leachables that will be used to generate the UF factors that must be applied to each detector. Two options exist for how UF values could be determined. The first method (conventional method) would be to develop a UF value independently for each detector without consideration of the detection capabilities of the other detectors. The goal in this approach would be to detect all compounds that respond at all on that detector using only that one detector. This is admittedly a difficult goal as it puts a substantial burden on that one detector to have universal response. It furthermore ignores the fact that some compounds do not respond at all on that detector (RF = 0) and assumes those compounds will be detected by another method without verifying or justifying that the strategy is capable. The difficulty in this approach can be seen in Table II by examining the LCMS positive ion RFs and noting that two of the compounds showed no response and the remaining three have a %RSD for the RFs of 114% indicating no sufficient UF can be calculated presumably resulting in the application of a UF of 10. In LCMS negative mode, three compounds were not detected and the remaining two compounds showed a % RSD of 75% and a required UF of 4. Similarly, the GCMS RF distribution showed a % RSD of 83% and required a UF of 6.1 and failed to detect three compounds. The combined strategy (LCMS positive and negative mode and GCMS with UF of 10, 4, and 6.1) failed to detect one compound (1,3,5-triphenylbenzene) as it had no response on the applied detectors. The second approach (multidetector approach) is based on determining the UF value in consideration of all of the detectors applied. This does not necessitate a consistent UF for all detectors but it is generally preferable as it minimize the need for sample concentration. The goal is to determine the most optimum group of detectors and methods that minimizes the required UF value needed to obtain the desired percent of coverage. Said another way, the goal is to demonstrate that a given combination of detectors, methods, and UF when applied in combination allow successful detection for an agreed upon percentage of compounds. The effectiveness of this approach can be seen in Table II by noting that a UF of 1 is sufficient to detect all five compounds so long as all detectors are considered (every compound has an RRF > 1 on at least one detector). The key assumption in the multidetector approach is that when a new extractable is analyzed, the probability of detection will be consistent with previous experience based on the analysis of a broadly constituted database of extractables. The underlying assumption in the multidetector approach can be justified based on data from a broadly constituted database of extractables and can be continuously improved as more extractables are analyzed by the method. It can further be verified in a particular analysis by analyzing standards of relevant chemistry and demonstrating suitable detection. In comparison, the conventional approach assumes that all nonresponding compounds not measured on one detector will be captured by the remaining detector without justification because these nonresponding compounds are not included in the UF determination for an individual detector and no reconciliation is typically performed to confirm the overall coverage of the approach using a broadly constituted database of extractables.
Practical Considerations When Applying a Multidetector AET Approach
A few practical considerations are also worth noting when applying the multidetector approach to the AET determination. First, it is important to remember that in all cases (using a multidetector or not) that appropriate surrogate standard selection is essential. This presumes a working knowledge of the method-specific RF distributions for each detector, which requires the creation of a RF database specific to each detector for a given method. As stated previously, this database should be broadly constituted (universal) so as to allow for detection of expected and unexpected extractables and leachables as examples exist of toxic leachables that would not have been predicted based on the materials of construction. Further verification of the methods appropriateness for the sample under study can be performed using chemically relevant standards as judged based on the materials of construction or early screening work as part of system suitability to confirm that expected extractables are well covered by the specific multidetector approach (combination of detectors, methods, and UF values applied).
In the multidetector approach, an evaluation is performed on each signal observed on any detector/chromatographic method and any compound that shows a signal above the AET on at least one detector must be included in the study. This by definition requires that an AET be established on each detector so that an evaluation can be made. It is the combination of all of these evaluations that must be sufficient to detect all relevant extractables and leachables as no one method could ever be expected to cover all compounds. The analysis presented here assumed that the surrogate standard used to set the AET had a response that was at the average of the RF distribution for each detector. This avoids bias caused by a strong or weak surrogate standard response. If a particularly strongly responding surrogate standard is selected, then this has the effect of increasing the threshold value and decreases the conservatism of the assessment. Similarly, if a poorly responding standard is selected, this decreases the magnitude of the threshold value making the study more conservative. In practice, it is difficult if not impossible to identify a single standard that would be at the center of the response distribution for multiple detectors. The different detection principles and classes of compounds to which they are best suited generally necessitates that different standards be applied for different detectors. Even in the limited case of a single detector, it is difficult to identify a standard that has a response that is at exactly the average of the RF distribution of even that one detector. Two methods can be proposed to account for the resulting distribution bias. The first would be to use a surrogate standard appropriate to each detector that is at or below the middle of the distribution for that detector. The extent to which the surrogate standard response is below the average response would further increase the conservatism of the study. If the response of the surrogate is higher than the average response, then a mathematic adjustment could be applied to increase the protectiveness of the threshold in proportion to the magnitude of the bias introduced. This could be accomplished using the RRF value for the surrogate standard and the average RRF for the distribution as determined for each detector and could be expressed mathematically as shown in eq 5:
 (6)
(6)
This may be preferred in cases for which a surrogate standard was selected before determining the distribution characteristics as both of these mechanisms can account for distribution bias. It is also noted that the use of the average peak area for a series of surrogate standards selected to be appropriate for each detector system could be substituted for a single standard to obtain a value for the threshold that approaches the average of the distribution on that detector. This approach has the benefit of simultaneously demonstrating the suitability of the method to detect compounds with a range of RFs (strong and weakly responding compounds) that can then serve as quantitation standards following identification. A set of standards should ideally be selected for each detector such that they have an average RRF near the average of the distribution on that detector.
This then brings up one final important consideration, which is that the sensitivity (limit of detection [LOD] or limit of quantification [LOQ]) for the various detectors must be sufficiently low to reach the AET. Detector sensitivity is not the same for all detectors. Thus, the ability to effectively apply a full multidetector approach is dependent upon the AET value required. If the AET is so low that at least one of the detectors cannot hit the required sensitivity, then the protectiveness of the study will be reduced and the UF may need to be increased to compensate. Fortunately, the magnitude of the UF can generally remain low so long as at least one orthogonal detector is applied in addition to GCMS and LCMS. The authors would also note that there is ongoing debate regarding the use of LOD or LOQ as the appropriate measure of sensitivity when applied to AET evaluation. The current ISO 10,993-18 guidance states “if one purpose of the analytical testing is quantification, the AET should be higher than or equal to the LOQ” (5). The AET was originally defined as “the threshold at or above which a chemist should begin to identify (emphasis added) a particular leachable and/or extractable and report it for potential toxicological assessment” (7). It is the authors’ opinion that although it is always preferred to have improved signal to noise in any evaluation (i.e., use LOQ), LOD is the appropriate limit given that the AET is a reporting threshold (compounds above this limit must be reported) and is a starting place from which to begin identification. Having a 3:1 signal/noise ratio (the common understanding of LOD) is sufficient to determine if something is “detectable as above the noise” hence it is a suitable metric for determining if a signal is above the AET. Quantitation is better reserved for a later stage in the analysis at which more options exist for improving method sensitivity, such as using targeted quantitation. This is an analytical necessity in cases in which instrumental/method LOQ is greater than the desired AET value and for which options to increase method LOQ have been exhausted. Using either LOD or LOQ does not fundamentally change the multidetector strategy so long as each detector can reach the required sensitivity limit. Finally, it is our opinion that the cost of increasing method sensitivity through sample concentration (3.3× increase in concentration factor needed for LOQ vs. LOD) and the resulting potential compound loss or degradation likely outweighs the benefit obtained by using LOQ at this early stage in the analysis.
Conclusion
The AET is an important tool for practical screening of organic extractables and leachables as it minimizes the amount of effort that must be expended identifying/quantifying compounds for which there is a very low risk of toxicologically adverse effects. As demonstrated by previous product recalls, screening methods for extractables and leachables should have broad detection capabilities as it is not always possible to predict which toxicologically relevant leachables may be present in a medical product. Successful application of the traditional AET approach is dependent on analytical responses being consistent, compound to compound, a circumstance that is not realized using the common detection methods applied for chromatographic screening for organic extractables and leachables. For poorly responding analytes, the AET may lose its ability to mitigate risk as the poorly responding analytes may appear to be below the AET based on response but are actually above the AET based on concentration.
To compensate for RF variation, the AET can be revised via the use of a UF that is related to the magnitude of the RF variation for the distribution of all extractables and leachables analyzed using the method. However, AET adjustment becomes problematic for certain individual analytical methods, such as LCMS, where RF variation is large and the UF correction becomes challenging, both mathematically and practically. These problems are substantially reduced, however, using a multidetector approach, an analytical approach that leverages multiple chromatographic methods with multiple complementary and orthogonal detection methods and a UF value verified to provide sufficient coverage using a broadly constituted database of extractables. The multidetector approach provides an alternative means to reduce the effects of RF variation through the application of a more optimum detector and thus reduce the number of unreported compounds present in a sample at or above the AET while also reducing the need for large UF. By extension, this reduces the need for additional sample preparation and concentration and with it the risks of loss or degradation of extractables. Thus although both methods (applying a larger UF and using a multidetector appproach) would in theory provide an equal degree of protection considering only response variation, the multidetector approach has the advantage of requiring less sample preparation and concentration to reach the same extent of coverage and provides a more protective result for cases in which sample preparation causes loss or degradation of extractables and leachables.
Appropriate surrogate standard selection and correction for distribution bias remains an important consideration no matter which strategy is applied. Standard and UF selection are integrally related and standards must be selected with a knowledge of the RF distribution for each detector. The use of chemically relevent standards could be used as a part of system suitability to validate the applied strategy for a particular device chemistry.
Based on the concepts that (a) use of multiple detectors makes it more likely that at least one detector will effectively respond to every extractable or leachable and (b) that the protectiveness of the AET is ensured for each compound so long as it can be properly applied on at least one detector (and not necessarily all detection methods simultaneously), this manuscript has established that a properly designed and appropriately complementary and orthogonal multiple detection strategy can ensure that the AET is protective without the use of large and largely unmanageable UFs.
Conflict of Interest Declaration
The authors have no conflicts of interest to disclose; however, Drs. Jordi and Heise note their affiliations with contract research organizations (CROs) that provide extractables and leachables services to the pharmaceutical, food contact, and medical device industries.
- © PDA, Inc. 2021
In This Issue




{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Introduction: The Analytical Evaluation Threshold
- AET Evaluation Using Multiple Orthogonal and Complementary Separation and Detection Methods
- Answers to Some Potential Critiques
- Practical Considerations When Applying a Multidetector AET Approach
- Conclusion
- Conflict of Interest Declaration
- References
- Figures & Data
- References
- Info & Metrics
Related Articles
Cited By...
- No citing articles found.