
Abstract
Burkholderia cepacia has recently received a considerable attention as one of the major risks in susceptible pharmaceutical products. This microorganism can easily propagate and cause vast and severe contamination, especially to the water supplies for pharmaceutical companies. Moreover, it proliferates within the products and can cause severe infections for humans. Therefore, fast and sensitive detection of these bacteria is of a great demand. The present study introduces improved application of a polymerase chain reaction assay with relatively high sensitivity and specificity for the direct detection of B. cepacia from the aqueous pharmaceutical products. A semi-nested polymerase chain reaction approach using the primer set BCR1/BCR2 followed by BCR1/Mr yielding a 465 bp fragment of the recA gene was applied and tested using both crude lysate from isolated colonies and DNA directly extracted from artificially prepared and spiked reference syrup. The polymerase chain reaction assay showed no interference with other bacterial reference and environmental strains tested, including Staphylococcus aureus ATCC® 6538, Pseudomonas aeruginosa ATCC® 9027, Escherichia coli ATCC® 8739, Salmonella abony NCTC® 6017, Bacillus subtilis ATCC® 6633, Micrococcus luteus, Staphylococcus warneri, Pseudomonas fluorescens, Pseudomonas putida, and Ralstonia pickettii. Moreover, this semi-nested assay showed a detection limit of around 10 colony-forming units per sample and could detect B. cepacia strains isolated from a municipal pre-treated potable water tank. Comparing the results for detection of B. cepacia in 100 randomly collected commercial syrup preparations using both conventional standard method and polymerase chain reaction assay revealed that B. cepacia was detected in two samples using polymerase chain reaction assay while all samples showed negative results by conventional culturing and biochemical methods. These results highlight the advantage of using this polymerase chain reaction assay to detect B. cepacia in contaminated pharmaceutical products and even water for pharmaceutical purposes, without the need of culturing or pre-enrichment, where it may give false-negative results and may be misidentified when biochemically tested.
- B. cepacia
- Susceptible pharmaceutical products
- Semi-nested polymerase chain reaction
- conventional culturing
- Biochemical methods
1. Introduction
Burkholderia cepacia (B. cepacia) is a pathogen that can infect immunocompromised populations, which include elderly people, pregnant women, young children, and people suffering chronic illness. However, B. cepacia is less frequently associated with illness in healthy non-immunocompromised patients (1).
B. cepacia complex (Bcc) comprises a group of related bacterial species widely distributed in nature and in artificial habitats (2). They are Gram-negative, motile, aerobic bacteria with non-fermenting properties. They live in nature especially in soils, water, and botanical products (3). They are multi-drug resistant organisms that can resist many disinfectants, cleansers, and antiseptics and is not affected by many preservatives. Bcc has also a great ability to form biofilms and contaminate plastics, metals, water systems, and consequently pharmaceutical facilities. There is a major risk to the pharmaceutical products and the patients when the process water used in pharmaceutical manufacturing is contaminated with Bcc (4).
Each pharmaceutical company is responsible for developing its own microbial specifications regarding the non-sterile products produced in its facility (5). The USP “Microbial Enumeration Tests” and “Tests for Specified Microorganisms” provide methodology for selected indicator microorganisms, but not all objectionable microorganisms (6, 7). It is well known that Bcc is objectionable if found in preparations to be inhaled or even administered locally using the nasal route as well as topicals used on broken skin. However, the USP chapters do not provide any detection or identification methods for B. cepacia (8).
According to the U.S. Food and Drug Administration (FDA) findings, Bcc had contaminated products even in the presence of one or more antimicrobial preservatives. The FDA faced an unusual case of contamination in that 10 lots of incoming solution passed initial release testing for bioburden. Later samples taken from these same lots of bulk solution showed failing levels of Bcc indicating that the bacteria was proliferating in the solution. Therefore, reliance on conventional testing of finished products has not been successful for detecting and eliminating Bcc contamination hazards (1, 9) However, the pharmaceutical manufacturer is responsible for controlling the drug manufacturing process to exclude potentially harmful microorganisms from entering the process. For this reason, research is needed to develop reliable methods for the detection of Bcc in pharmaceutical products, especially given that Bcc has the potential for nutrient shock during conventional cultivation, giving false-negative results (1).
Although taxonomy studies of B. cepacia have improved its identification (10), differentiation of B. cepacia species from other related taxa, such as Ralstonia, Cupriavidus, Pandoraea, Achromobacter, Brevundimonas, Comamonas, and Delftia species, remains difficult (11).
Numerous advances have been made in the identifications of B. cepacia using traditional and molecular techniques; however, the great diversity among B. cepacia strains limited the utility of the methods applied (12). Among the molecular methods used for identification of B. cepacia is the recA gene–based analysis, which was found to be effective for species identification based on 94–95% similarity of the recA gene between different Bcc species and 98–99% similarity within Bcc species (13). Moore et al. (14) have previously developed a semi-nested polymerase chain reaction (SN-PCR) method based on recA gene amplification for the direct identification of B. cepacia from sputum of patients with cystic fibrosis with a sensitivity reaching 101 and 102 cfu/g (colony-forming units per gram) sputum for genomovars IIIa + b and genomovar II, respectively.
Taking into consideration the low level of bioburden that might contaminate aqueous pharmaceutical products during manufacturing and the need for a sensitive method for detection of B. cepacia in susceptible products, the aim of the present work was to optimize and investigate the applicability of a SN-PCR method based on recA gene amplification, which was previously applied for the detection of B. cepacia from sputum samples of cystic fibrosis patients (14), for the detection of B. cepacia in pharmaceutical aqueous syrup preparations without previous culturing or enrichment and to compare the optimized method with conventional methods that are mostly followed in the microbiological control laboratories of the pharmaceutical manufacturing facilities. B. cepacia ATCC 25416 and environmental B. cepacia isolates from a municipal potable water tank were used as reference strains to represent the species that can most probably contaminate aqueous pharmaceutical preparations.
2. Materials and Methods
Unless otherwise specified, all tests were conducted under aseptic conditions and in triplicate.
2.1. PCR-Based Detection Methods
2.1.1. DNA Extraction and Purification:
DNA extraction and purification steps were performed using a QIAamp® DNA Mini kit, except for sensitivity and commercial products testing, where we used a QIAamp® DNA Blood Midi kit. DNA purification from bacterial colonies or from inoculated syrup preparations was performed according to the manufacturer's instructions. All protocols followed were spin protocols and the centrifugation steps were carried out using a Corning® LSETM high-speed micro centrifuge device for the QIAamp® DNA Mini kit and Corning® LSE™ compact centrifuge for the QIAamp® DNA Blood Midi kit.
After extraction, the solution containing DNA was measured for DNA content using Thermo Scientific™ NanoDrop, and the volume containing 100–200 ng (∼150 ng) of DNA was used in PCR reaction.
2.1.2. PCR Reactions Mix:
The PCR reaction mix for all implemented PCR reactions was composed of 12.5 μL Qiagen® Taq PCR master mix kit (equivalent to 1 X concentrate), 1 μL of 25 mM MgCl2 (equivalent to 2.5 mM MgCl2 in the total reaction), 0.5 μL Primer 1, 0.5 μL Primer 2 (equivalent to 0.2 mM from each primer in the total reaction), 1–5 μL DNA template and complete to 25 μL using Qiagen® RNAse free water.
2.1.3. Semi-Nested Polymerase Chain Reaction (SN-PCR) Detection Method:
A SN-PCR method employing the primer set BCR1/BCR2 followed by primer set BCR1/Mr (Table I) generating an amplicon of 465 bp as previously described (13, 15) was carried out. This method was applied on the following standard microorganisms: Staphylococcus aureus ATCC® 6538, Pseudomonas aeruginosa ATCC® 9027, Escherichia coli ATCC® 8739, Salmonella abony NCTC® 6017, Bacillus subtilis ATCC® 6633, and B. cepacia ATCC® 25416. Additionally, the following environmental isolates were recovered from an environmental monitoring program of a pharmaceutical facility and identified using a Vitek 2 compact identification system: Micrococcus luteus, Staphylococcus warneri, Pseudomonas fluorescens, Pseudomonas putida, and Ralstonia pickettii. DNA was extracted from bacterial colonies of the previously mentioned microorganisms. SN-PCR was carried out in two successive rounds, where 2 μL of the PCR product from the first round was employed as DNA template for the second round, and the thermal cycling conditions were as follows: the first round was 96 °C for 5 min, followed by 25 cycles at 96 °C for 1 min, 58 °C for 1 min, 72 °C for 2 min, followed by a final extension at 72 °C for 10 min. The second round was 96 °C for 5 min, followed by 30 cycles at 96 °C for 5 min, 60 °C for 1 min, 72 °C for 1 min, followed by a final extension at 72 °C for 10 min using a GeneAmp®-PCR system 9700 - Applied Biosystems. PCR products were detected by electrophoretic separation using a Biometra® XS/S electrophoresis system on 1.5 % Ultrapure Invitrogen® agarose gel and a Qiagen® GelPilot 100 bp Plus ladder at 100 V for 30–45 min. The gels were visualized and photographed using a Life Technologies E-gel imager®. Also, 23 environmental isolates from a municipal pre-treated water tank were tested using SN-PCR assay by extracting DNA from the bacterial cells and previously were biochemically identified using a Vitek® 2 compact identification system.
List of Primers Used for SN-PCR
2.2. Test Samples
Five liters from an aqueous syrup preparation was prepared as a reference, sterilized using 0.22 μm membrane filters, and stored and refrigerated to simulate commercial aqueous syrup samples within the whole study. The reference preparation composed of the following: purified water, Quinoline Yellow (D&C Yellow NO10), ethyl alcohol, methyl paraben, Plasdone K25 (PVP25) (Povidone K25), propyl paraben base, sorbitol 70/70% solution, sucrose, and a flavor (Orange Flavour E9904557). Further, 100 randomly collected commercial aqueous preparations were included in order to be used during the application of the testing procedures. The commercial preparations were labeled from P1 to P100.
2.3. Conventional Detection Methods
Conventional method suitability testing was carried out according to the procedures of the recovery in liquid medium by the membrane filtration technique in the USP (16⇓–18). Two dilutions were prepared from the reference preparation and from each of the commercial products (1:10 and 1:100) using USP phosphate-buffered solution (PBS) + 4% Tween® 80 as a diluent.
The microorganisms used in conventional method suitability testing were as follows: Staphylococcus aureus ATCC® 6538, Pseudomonas aeruginosa ATCC® 9027, Bacillus subtilis ATCC® 6633, Candida albicans ATCC® 10231, Aspergillus brasiliensis ATCC® 16404, and B. cepacia ATCC® 25416 to ensure that the used methods are highly reliable in the recovery of various microorganisms.
Three groups each of 200 mL from the reference preparation and from each of the commercial products were pooled in sterilized bottles.
For each preparation, a 10 mL sample from each of the 1:10 and 1:100 dilutions was inoculated with 0.1 mL containing <100 cfu of each microorganism. Then samples were then filtered through membrane filters of 0.45 μm pore size, rinsed once with 100 mL PBS 4%. The membrane filters were aseptically transferred to tryptic soy agar (TSA) plates for recovery of bacteria and Saboraud dextrose agar (SDA) plates for recovery of Candida albicans and Aspergillus brasiliensis. A negative control was carried out for each dilution from each bottle used from the reference preparation or commercial products. The microbial recovery of each dilution for each microorganism used was compared to a positive control containing the microorganism and the media without the product. The lowest dilution factor showing a microbial count greater than or equal to 50% of the positive control of each microorganism was the accepted dilution. For recovery of B. cepacia, another membrane filter was aseptically transferred to 100 mL tryptic soy both supplemented with 4% Tween® 80 (TSB 4%), incubated at 30–35 °C for 18 h, and then 0.1 mL from the incubated TSB 4% bottles was streaked on TSA, cetrimide, and BBL™ oxidation/fermentation-polymyexin-bacitracin-lactose (OFPBL). After incubating the TSA and cetrimide plates at 30–35 °C for 18 h and OFPBL media at 30–35 °C for 4 days, growth was recorded and the lowest dilution showing growth and a microbial count greater than or equal to 50% of the positive control was chosen as the accepted dilution.
Each of the reference and the commercial samples was tested later using the accepted dilution.
2.4. Testing of Filterable Pharmaceutical Preparations via SN-PCR
One hundred milliliters of the reference syrup were spiked with B. cepacia ATCC 25416 (on triplicate basis) and with the environmental isolates that were identified as B. cepacia either biochemically or by SN-PCR at an inoculum size of 107 cfu/100 mL. Bacterial DNA was extracted by filtering the whole spiked dilution through a membrane filter. The membrane filter was soaked in 5 mL saline for 30 min, and then bacterial genomic DNA was extracted from the saline solution using a QIAamp® DNA Mini kit, following the manufacturer's instructions, and was used as a template in the first round of the SN-PCR for identification of B. cepacia.
2.5. Sensitivity of SN-PCR versus Conventional Methods
B. cepacia ATCC 25416 was serially diluted to 1000 ± 200 cfu/0.1 mL, 100 ± 20 cfu/0.1 mL, and 50 ± 10 cfu/0.1 mL. An aliquot of 0.1 mL from each serial dilution was used to spike 10 of the accepted dilutions identified by the conventional method suitability testing of the prepared syrup. Each of the spiked dilutions was tested by membrane filtering of the whole volume, transferring the membrane filters on TSB 4%, incubated at 30 - 35 °C for 24 h, and then streaking 0.1 mL on TSA, cetrimide agar, and OFPBL agar.
Ten groups each of 100 mL of the prepared syrup (without being diluted) were spiked with 0.1 mL containing around 10 cfu (10–15 cfu) of B. cepacia ATCC 25416 as counted on OFPBL agar, then the bacterial DNA was extracted and purified directly from the product, as previously described, with the following modifications of using 5 mL Qiagen® tissue lysis (ATL) buffer for soaking the membrane filters, using Qiagen® proteinase K instead of Qiagen® protease, incubating the sample with ATL buffer and proteinase K at 56 °C for 3 h before adding Qiagen® lysis (AL) buffer, warming Qiagen® elution (AE) buffer to 60 °C, and following the steps of purification of DNA from whole blood using the QIAamp® Blood Midi kit (Spin Protocol). During the elution phase, 100 μL of the warmed AE buffer was added to the QIAamp® Midi column, left for 10 min before starting elution centrifugation, re-elution with the same eluate, and finally elution with another 100 μL of the AE buffer. The final eluate was evaporated to around 50 μL using a Thermoscientific® heat block at 60 °C with frequent vortexing to increase the DNA concentration in the eluate, and then SN-PCR method was applied as previously described.
2.6. Testing Commercial Products by SN-PCR and Conventional Methods
One hundred aqueous commercial products collected randomly from the market were tested by both conventional methods and SN-PCR for the detection of B. cepacia. One hundred milliliters of each product was used, half of the product's volume was diluted to the suitable dilution factor and was membrane-filtered, and the membrane was rinsed with 100 mL 4% PBS. The membrane filter was then aseptically transferred to TSB 4% and incubated at 30–35 °C for 24 h. One hundred microliters of the incubated TSB 4% was then streaked on TSA, cetrimide agar, and OFPBL agar. All recovered colonies were identified via biochemical reactions using a Vitek 2 compact system. The other half of each product was filtered undiluted, and the same steps of SN-PCR in sensitivity tests were followed.
3. Results
3.1. SN-PCR Detection Method
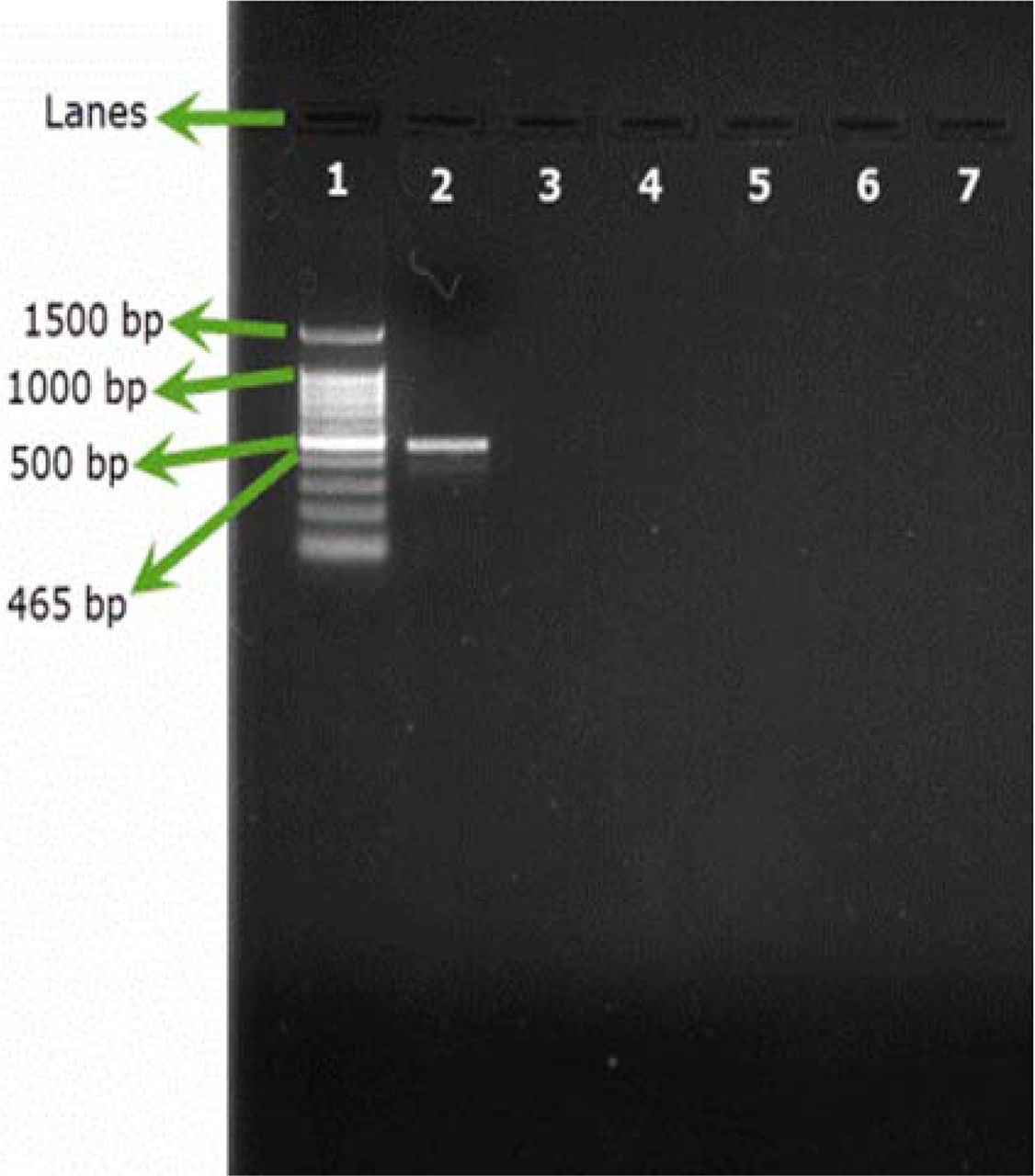
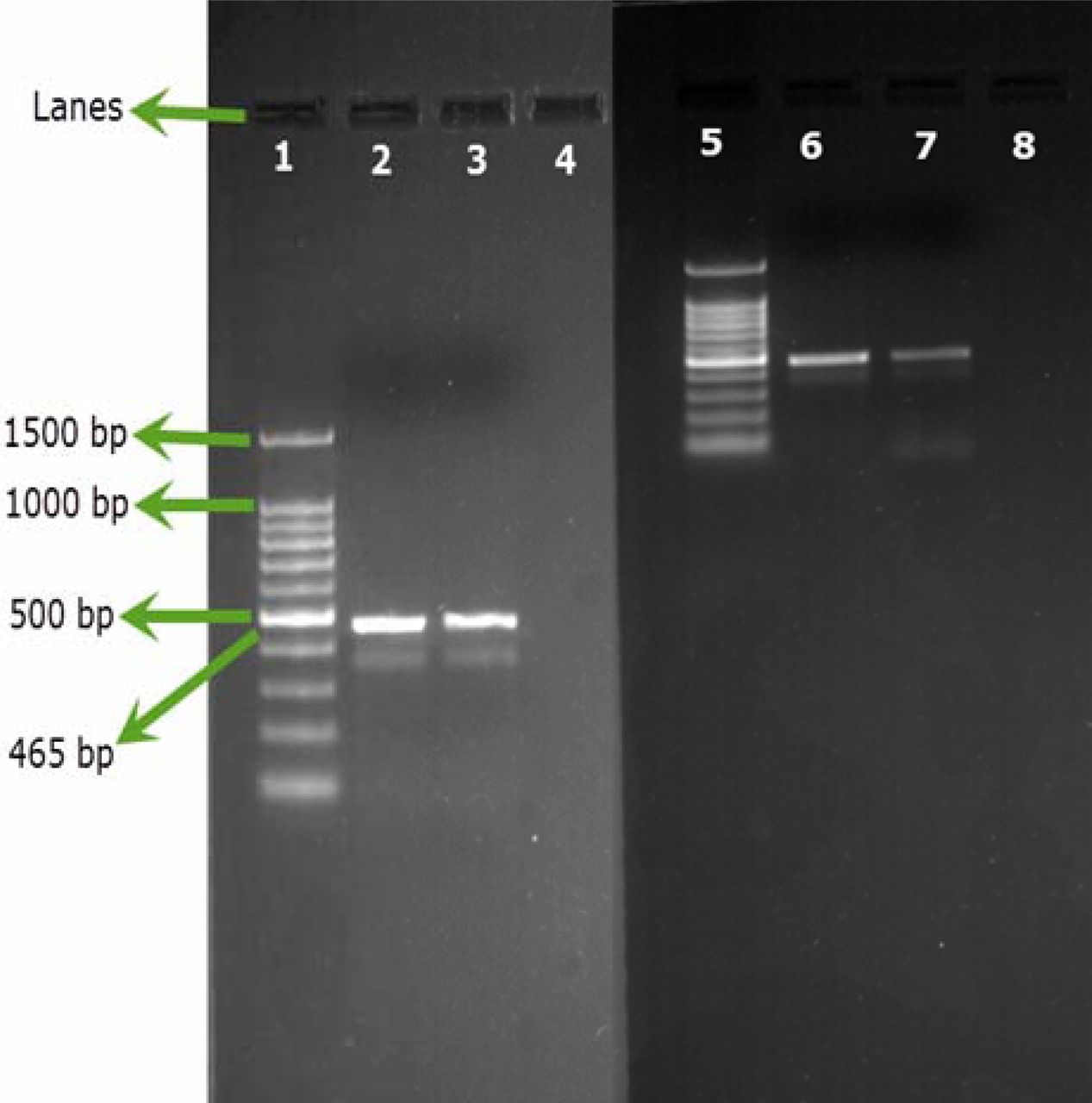
Only B. cepacia ATCC 25416 gave positive results and showed the characteristic band at 465 bp, while other reference strains and environmental isolates showed negative results (Figures 1A, 1B). Screening of 23 environmental isolates from a municipal potable water tank using SN-PCR and the Vitek® 2 compact identification system revealed that only four out of the 23 isolates were identified as B. cepacia using the Vitek® 2 system, while six showed positive results for B. cepacia using SN-PCR (Table II) including those identified as B. cepacia using the Vitek® 2 system.

Semi-nested PCR (SN-PCR) detection method using reference strains. Lane 1: Qiagen® GelPilot 100 bp Plus ladder, Lane 2: B. cepacia (ATCC 25416), Lane 3: Staphylococcus aureus ATCC® 6538, Lane 4: Pseudomonas aeruginosa ATCC® 9027, Lane 5: Escherichia coli ATCC® 8739, Lane 6: Salmonella abony NCTC® 6017, Lane 7: Bacillus subtilis ATCC® 6633, Lane 8: negative control.

Semi-nested PCR (SN-PCR) detection method using environmental strains. Lane 1: Qiagen® GelPilot 100 bp Plus ladder, Lane 2: B. cepacia (ATCC 25416), Lane 3: Micrococcus luteus, Lane 4: Staphylococcus warneri, Lane 5: Pseudomonas fluorescens, Lane 6: Pseudomonas putida, Lane 7: Ralstonia picketti.
Identification of Environmental B. cepacia Isolates by SN-PCR and Vitek 2 Compact Identification System
3.2. Conventional Detection Methods
The triplicate samples of the 1:10 dilution of the prepared reference syrup showed successful count recovery for all the microorganisms tested and showed the expected positive growth on TSA, cetrimide agar, and OFPBL agar for B. cepacia, which was successfully identified using the Vitek® 2 compact identification system. Ninety-eight of the 1:10 dilution of the commercial samples showed the expected recovery and B. cepacia–positive results and only two preparations (P45 and P57) showed the accepted results with the 1:100 dilution.
3.3. Testing of Filterable Pharmaceutical Preparations via SN-PCR
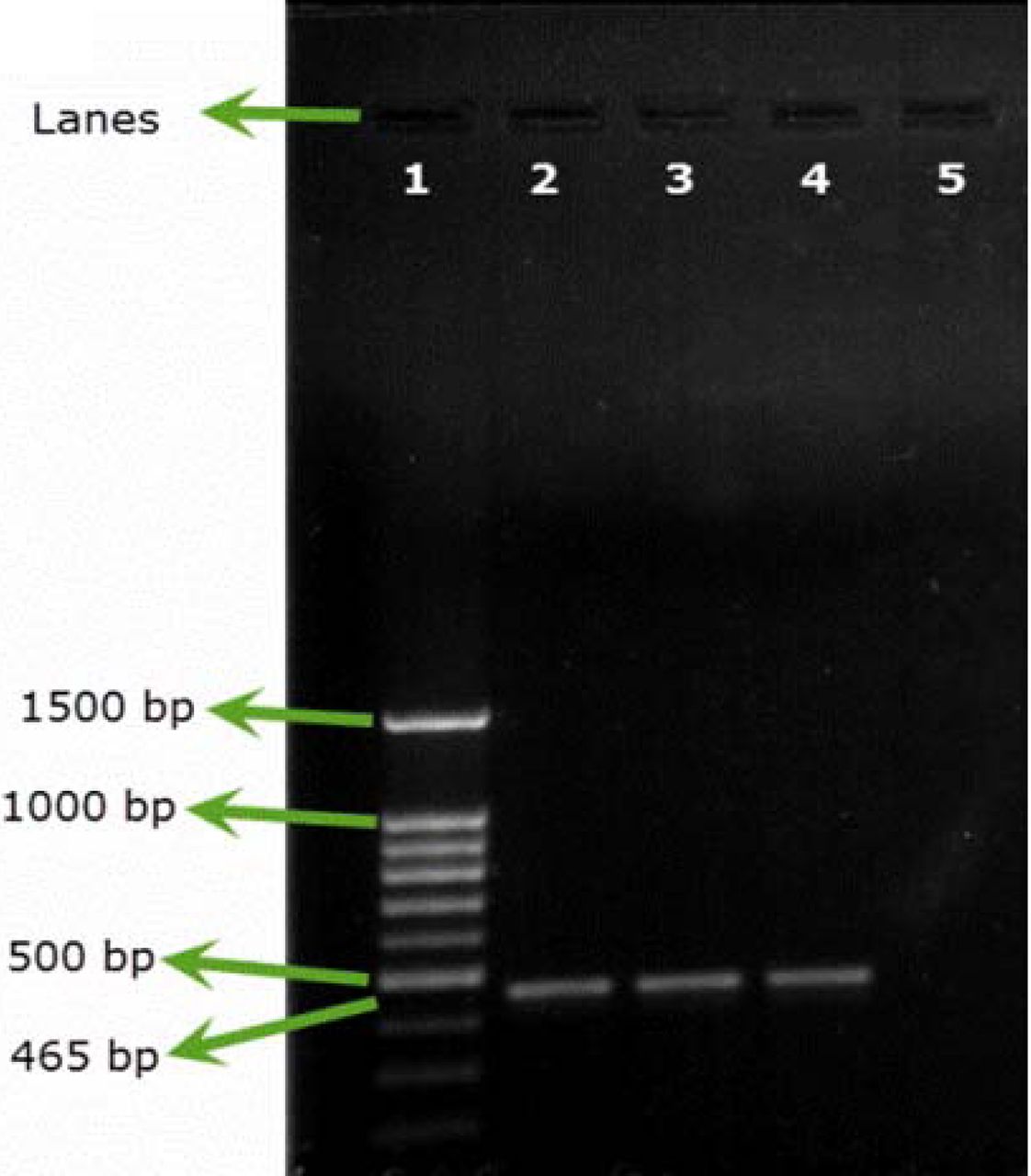
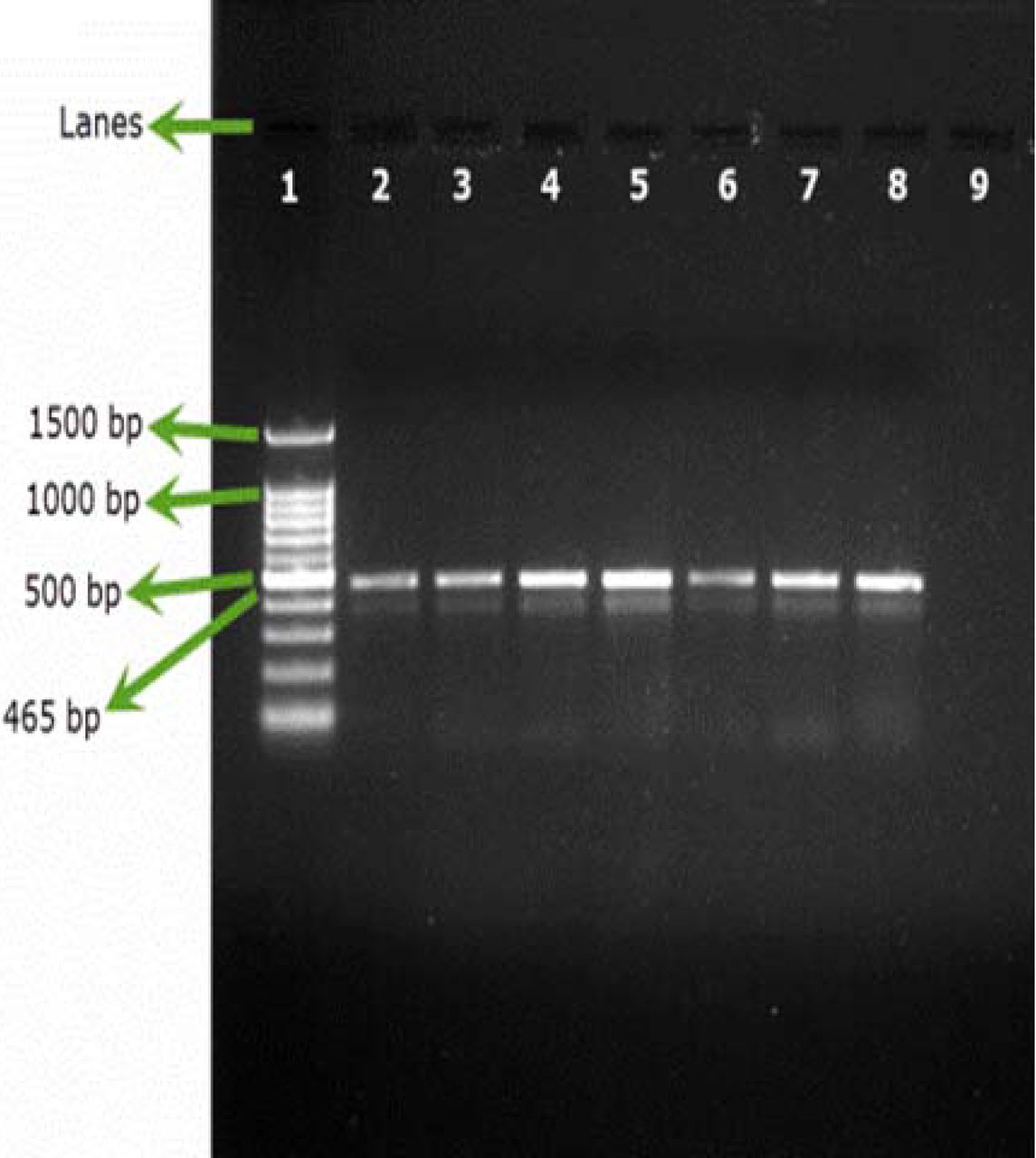
All the samples inoculated either with B. cepacia ATCC or with environmental B. cepacia isolates showed positive results using SN-PCR directly from the products without the need for pre-enrichment steps (Figures 2A, 2B).

Testing artificial filterable pharmaceutical preparation via SN-PCR using B. cepacia (ATCC 25416) at an inoculum size of 107 cfu/100 mL. DNA was extracted from the preparation. Test was repeated 3 times. Lane 1: Qiagen® GelPilot 100 bp Plus ladder, Lane 2: pharmaceutical preparation containing B. cepacia (ATCC 25416) 107 cfu/100 mL, Lanes 3 and 4: repeated test, Lane 5: negative control.

Testing artificial filterable pharmaceutical preparation via SN-PCR using 6 environmental strains of B. cepacia. DNA was extracted from the preparation. Lane 1: Qiagen® GelPilot 100 bp Plus ladder, Lane 2: pharmaceutical preparation containing B. cepacia (ATCC 25416) 107/100 mL, Lanes 3–8: environmental isolates (1–6), Lane 9: Negative control.
3.4. Sensitivity of SN-PCR versus Conventional Methods
All the samples spiked with either 1000 ± 200 cfu/0.1 mL or 100 ± 20 cfu/0.1 mL showed positive results on TSA, cetrimide agar, and OFPBL agar (Tables IIIA, IIIB). The samples spiked with 50 ± 10 cfu/0.1 mL and tested conventionally via normal culturing methods showed five negative results, as shown in Table IIIC. No further dilutions were used due to the negative results shown.
Sensitivity of Conventional Methods for Detection of B. cepacia Using Inoculum Sizes Ranging from 50 to 1000 cfu/0.1 mL
B. cepacia Inoculum Size 1000 ± 200 cfu/0.1 mL
B. cepacia Inoculum Size 100 ± 20 cfu/0.1 mL
B. cepacia Inoculum Size 50 ± 10 cfu/0.1 mL
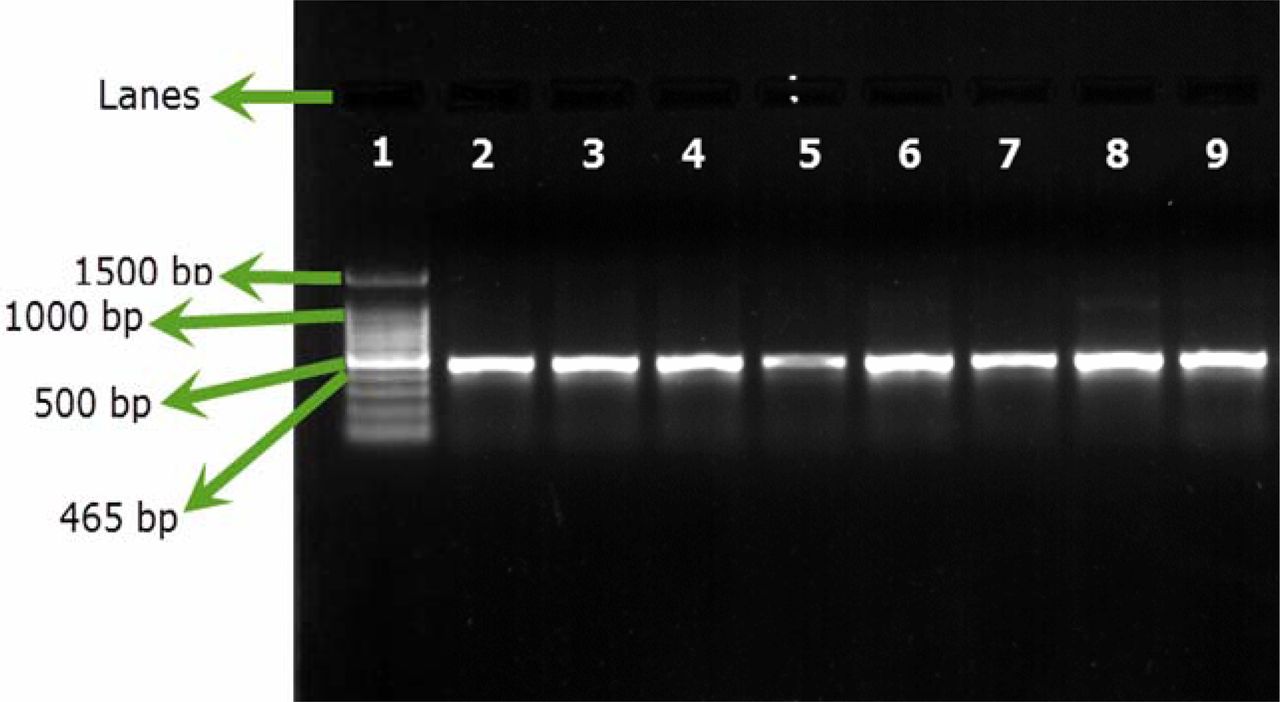
All the samples spiked with approximately 10 cfu showed positive results for B. cepacia upon testing via SN-PCR as shown in (Figure 3), presenting the results of 7 groups out of the 10 spiked groups.

Sensitivity of SN-PCR for testing artificial filterable pharmaceutical preparations using B. cepacia (ATCC 25416) at an inoculum size of 10 cfu/preparation. Test was repeated 10 times. Lane 1: Qiagen® GelPilot 100 bp Plus ladder, Lane 2: B. cepacia (ATCC 25416) DNA extracted from colony, Lane 3: pharmaceutical preparation containing B. cepacia (ATCC 25416) 10 cfu/preparation, Lanes 4–9: test repetitions.
3.5. Testing Commercial Products by Both Conventional Methods and SN-PCR
No aqueous commercial products showed positive results for B. cepacia when tested conventionally, while two products, P57 and P82, showed positive results for B. cepacia when tested via SN-PCR (Figure 4).

Testing commercial products by SN-PCR. Lane 1: Qiagen® GelPilot 100 bp Plus ladder, Lane 2: B. cepacia (ATCC 25416) (DNA extracted from colony), Lane 3: Product #57, Lane 4: negative B. cepacia product, Lane 5: Qiagen® GelPilot 100 bp Plus ladder, Lane 6: B. cepacia (ATCC 25416) (DNA extracted from colony), Lane 7: Product 82, Lane 8: negative B. cepacia product.
4. Discussion
B. cepacia is an opportunistic pathogen that causes diseases primarily among immunocompromised populations. Among the most serious conditions caused by B. cepacia are pneumonia, bacterial infection that occurs in patients with impaired immune systems, and chronic lung disease, particularly cystic fibrosis (18).
B. cepacia are among the most antimicrobial agent-resistant organisms. They have the ability to grow in low-nutrient conditions (19) and in the presence of chemical preservatives (20). Because the most common source of contamination is water, aqueous products are especially at risk because of B. cepacia's ability to remain viable in harsh conditions (1).
Because the organism may grow poorly or not at all when transferred from water (aqueous) systems to high-nutrient culture media, testing of finished product by conventional methods can produce misleading, false-negative results (1). Although the use of certain specialized, low-nutrient media could provide better results, the use of molecular methods for identification remains better.
In our study, we aimed at investigating and optimizing the detection of B. cepacia from aqueous pharmaceutical products using SN-PCR. We first examined the specificity of SN-PCR using DNA from a selected panel of microorganisms, representing reference indicator strains specified in USP under “Tests for Specified Microorganisms” and environmental strains isolated from an environmental monitoring program at a pharmaceutical facility and which would be likely to contaminate pharmaceutical products. Our results demonstrated that neither of the strains was amplified by SN-PCR. Previous studies have shown the specificity of the recA primers to amplify Bcc organisms (15). In addition, Moore et al. (14) confirmed the specificity of the primers against several species that may colonize the airways of cystic fibrosis patients. We further showed the ability of SN-PCR to detect B. cepacia strains isolated from a municipal pre-treated potable water tank that were not detected using the Vitek® 2 identification system. To investigate the applicability of SN-PCR to detect B. cepacia in pharmaceutical products, we spiked the accepted dilution of a reference pharmaceutical preparation with an inoculum of 107 cfu/mL of B. cepacia ATCC strain and environmental B. cepacia separately, with subsequent filtration and extraction of DNA from membrane filters using a commercially available DNA extraction kit. SN-PCR could detect B. cepacia in all spiked samples. For testing the detection limit of the method, we adopted a modified method for DNA extraction from biological fluids that maximized the DNA yield from pharmaceutical preparations, and we could detect around 10 cfu/preparation. In contrast, the conventional method could detect B. cepacia in the accepted dilution factor of the preparations spiked with 1000 and 100 cfu but showed five negative results in those spiked with 50 cfu. Finally, we applied the conventional method and the optimized method for the testing of 100 randomly collected commercial preparations. SN-PCR could detect B. cepacia in two pharmaceutical preparations that the conventional method failed to detect B. cepacia in.
5. Conclusion
SN-PCR is a reliable method that can be used in microbiological quality control laboratories in pharmaceutical facilities for the detection of B. cepacia from aqueous filterable pharmaceutical preparations with high specificity and sensitivity, allowing the detection of the organism in small quantities equivalent to around 10 cfu in the whole preparation, which helps to prevent false-negative results and possible proliferation of the microorganism in aqueous pharmaceutical preparations during shelf life. Further work will be done to test a higher number of commercial preparations using the optimized method.
Conflict of Interest Declaration
The authors declare that they have no competing interests.
- © PDA, Inc. 2016
References




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Identification of Burkholderia cepacia Complex by PCR: A Simple Way
- Design, Development, and Validation of a Culture-Independent Nucleic Acid Diagnostics Method for the Rapid Detection and Quantification of the Burkholderia cepacia Complex in Water with an Equivalence to ISO/TS 12869:2019
- Burkholderia cepacia Complex Bacteria: a Feared Contamination Risk in Water-Based Pharmaceutical Products
- Real-Time PCR Detection of Burkholderia cepacia in Pharmaceutical Products Contaminated with Low Levels of Bacterial Contamination